
Abstract
Molecular markers serve to assign individual samples to specific groups. Such markers should be easily identified and have a high discrimination power, being highly conserved within groups while showing sufficient variability between the groups that are to be distinguished. The availability of a large number of complete genomic sequences now enables the informed selection of genes as molecular markers based on the observed patterns of variability. We derived a new scoring system based on observed DNA polymorphic differences, and which uses the Bayes theorem as adapted by Wilcox. For validation, we applied this system to the problem of identifying individual species within a prokaryotic (
Background
Molecular methods to assign biological samples to specific groups (eg, taxonomic groups) have largely replaced morphological comparisons, allowing hundreds or even thousands of characters to be compared across samples.
1
Historically, numerous DNA-based approaches encompassing random whole-genomic analysis have been used to discriminate groups of organisms. These include methods like, among many others, restriction fragment length polymorphism (RFLP), or random amplification of polymorphic DNA (RAPD).2,3 Alternatively, sequences from genes, usually selected by their conserved, housekeeping roles, can be used.
2
However, it is often the case that existing markers provide insufficient resolution or are confounded by homoplasy, homologous recombination and lateral gene transfer.4,5 In recent years, thanks to great advances in sequencing technologies,6,7 the number and diversity of completely sequenced genomes is growing exponentially. This provides the basis for optimizing the selection of marker genes based on the analysis of the whole genetic complement of a given set of organisms. Earlier attempts to use whole-genome information to select marker genes that could best serve as predictors of phylogenetic relatedness include the use of scores based on the level of sequence identities from whole-genome alignments,
8
or the selection of unique sequence signatures present in a few species.
9
These methods, however, do not exploit the information from sequence variability within a species. Here we propose and evaluate an alternative algorithm for the selection of optimal genetic markers, which is based on the comparison of complete genomes. In brief, the basis of our strategy is to rank different genes according to the level of DNA polymorphism within and between defined taxonomic groups. More specifically, DNA polymorphism is measured as the average number of nucleotide differences per site,
10
and a conditional probabilistic statistic based on Bayes's Theorem as adapted by Willcox
11
is used to prioritize genes, so that genes presenting higher levels of polymorphism between groups but lower variation within a group receive higher scores. In order to validate the methodology, we apply it to the problem of selecting marker genes for the identification of individual species within a prokaryotic (
Methods
Sequence data
Complete genome sequences were downloaded from the National Center of Bioinformatics Information (NCBI) in Genbank (.GBK) format. These were: (1) chromosome I from the following Vibrio species and strains:
Alignments, polymorphism analysis, and molecular marker score calculation
Genome sequences mentioned above were divided into four different groups: (1) VibrioDS, containing only one representative genome for each Vibrio species, using the
Algorithm
The Bohle-Gabaldón (BG) score calculation is based on the level of DNA polymorphism in the Distinct Species (
If molecular marker with specific size is required (
BG score using DNA polymorphism (less than 4 genomes):
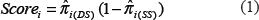
Scoring using DNA polymorphism and Size (less than 4 genomes)

Scoring using DNA polymorphism and Tajima's

Scoring using DNA polymorphism, Tajima's

The maximum value for Score is 1 using
Experimental validation analysis
Additional
DNA purification and amplification
Genomic DNA from
Molecular marker discrimination power analysis
To prioritize the markers, we developed a simple Discrimination Power (DP) score (5) based in Bayes's Theorem adapted by Willcox
11
which evaluates the maximum identity (Δ
Results
Automated prioritization of marker genes
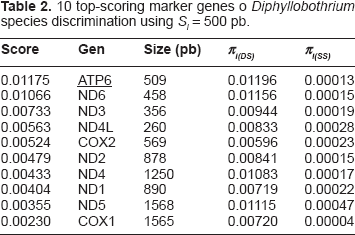
10 top-scoring marker genes for

10 top-scoring marker genes o
Experimental Validation
In order to validate the effectiveness of our approach we amplified these marker genes from additional strains of known taxonomic assignment but with no current genomic sequences available. The effectiveness of the markers, as measured by the Discrimination Power score (DP) described above, was compared to that of common markers used previously for these species. These were
Prokaryotic molecular markers genes comparison using Discrimination power scoring.

Eukaryotic molecular markers genes comparison using Discrimination power scoring.

Discussion
We have proposed and validated a novel approach for the informed selection of marker genes based on the observed levels of DNA polymorphism 10 among whole genomic sequences. Our results indicate that our approach effectively selects marker genes for species differentiation. Besides having greater discrimination powers than traditional markers, our markers also reduced the number of species that showed identical sequences for the marker. Nevertheless, in both genera studies, there are still some species that are too closely related to be differentiated with a single marker. The use of a combination of markers, or the selection of specific markers for that group of species within the genus would be required. Our approach has some minimal requirements. For instance, if the goal is to obtain marker genes for species differentiation in a given genus, a minimum of three different strain genomes belonging to two different species within the genus is required. Moreover, the design of primers may present problems if the sequences are too divergent, although this problem is shared with other approaches.
Our approach and scoring system method provides a new, powerful tool for the exploitation of available genome sequences to assist in the selection of marker genes. In both the eukaryotic and prokaryotic genera tested, the theoretical analyses showed excellent correlation with empirical results and showed a better performance than molecular markers previously proposed by different authors for the same species. The adaptation of Bayes theorem permitted the use of a conditioned statistic that prioritizes genes showing low DNA polymorphism inside the same species (different strains), while displaying high DNA polymorphism between different species.
Author Contributions
Conceived and designed the experiments: HB, TG. Analysed the data: HB, TG. Wrote the first draft of the manuscript: HB, TG. Contributed to the writing of the manuscript: HB, TG. Agree with manuscript results and conclusions: HB, TG. Jointly developed the structure and arguments for the paper: HB, TG. Made critical revisions and approved final version: HB, TG. All authors reviewed and approved of the final manuscript.
Footnotes
Supplementary Data
The scoring system and the necessary re-formatting scripts have been implemented in PERL. The PERL scripts (SCORE. pl and XMFA.pl) and a user manual for Windows, Linux and Mac are available at http://www.bioinformatics.cl.
Acknowledgements
We would like to thank Dr. Bruno Gomez-Gil for the donation of fixed biological material from different
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.