
Abstract
Hypotheses about horizontal transfer of antifreeze protein genes to ice-living diatoms were addressed using two different statistical methods available in the program Prunier. The role of diversifying selection in driving the differentiation of a set of antifreeze protein genes in the diatom genus
Introduction
Genes coding for antifreeze proteins (AFPs) have been discovered in various taxa that occur in cold temperatures; these include unicellular eukaryotes, plants, bacteria, fungi, fish, crustaceans and insects.1–9 Two major groups of AFPs are recognized. One type prevents cell/body fluids from freezing while the other kind consists of proteins that make it possible for organisms to survive cell/body fluid freezing.1,5 It is thought that both AFPs and protein ice nucleators play an important role in both freeze avoidance and tolerance.1,5 The former prevents freezing while the latter limits supercooling and induce freezing. 1 In some prokaryotes and eukaryotes that survive cell/body fluid freezing, the AFPs act as cryoprotectants. Even though this process is not fully understood, it may take place through ice recrystallization inhibition and possibly also by cell membrane stabilization.5,14 Those organisms that survive through freeze avoidance have AFPs that lower the freezing temperature of body fluids noncolligatively without affecting the melting temperature.1,5
Genes coding for proteins with antifreeze properties have probably evolved independently in a number of lineages
10
while some organisms have most likely acquired the genes through horizontal gene transfer (HGT).4,11,12 The evolution of prokaryote genomes is thought to have been profoundly influenced by lateral gene transfer.
13
The effect of this process on eukaryote genome evolution appear to be much more prevalent than previously thought. This is evidenced by an increasing number of documented HGTs in eukaryotes, particularly in unicellular eukaryotes.13,14 For instance, the nuclear genomes of the diatoms
All the currently known AFPs in ice-living diatoms appear to act as cryoprotectants.4,11 AFPs, which show similar amino acid sequences to those found in diatoms, have also been described for different prokaryote, fungal and crustacean lineages.4,11 This observation suggests that diatoms may have acquired the AFP genes from distantly related taxa through HGTs.4,11 However, another possibility, which cannot completely be ruled out, is convergent evolution of AFP genes in diatoms and certain prokaryote, fungal and crustacean lineages.4,11
In this study, hypotheses about horizontal transfer of AFP genes to sea-ice living diatoms from distantly related taxa were tested using the program Prunier—an algorithm developed by
18
for inferring HGT events based on statistical criteria. The role of diversifying selection in driving the differentiation of a set of duplicated AFP genes in the diatom genus
Material and Methods
Sequences
The choice of taxa in the current study was largely influenced by the paper of,
11
that is one of the major goals was to test hypotheses of HGT using the taxa shown in Figure 2 in
11
Moreover, amino acid sequences similar to the various AFPs of the two
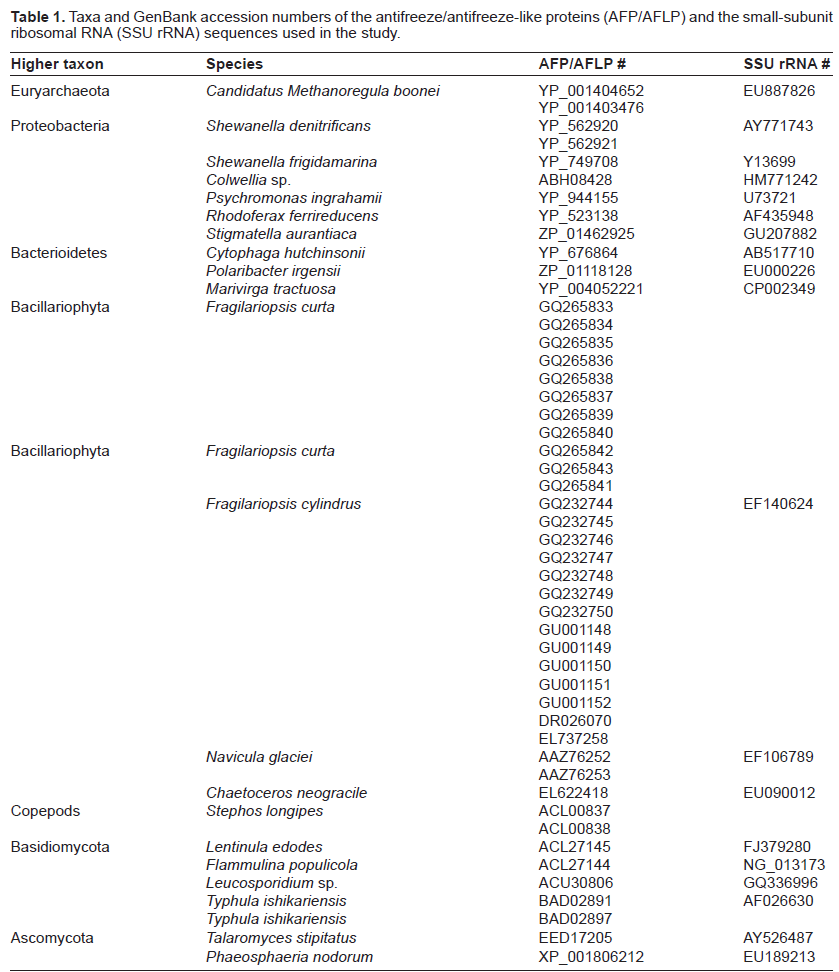
Taxa and GenBank accession numbers of the antifreeze/antifreeze-like proteins (AFP/AFLP) and the small-subunit ribosomal RNA (SSU rRNA) sequences used in the study.
Data analyses
The software package DAMBE (Version 5.0.5), 21 was implemented to manage the data and to match the codons against the aligned amino acid sequences. The alignments of the amino acid and SSU rRNA sequences, which are available upon request, were generated using the MAFFT (Version 6 22 ) Web Server available at http://mafft.cbrc.jp/alignment/server/. The alignment mode G-INS-i 23 with an offset value of 0.1 and the MAFFT homolog function “turned on” (all the other parameter settings were default values), was used for the proteins. When aligning the SSU rRNA sequences option Q-INS-i 24 was implemented. Only the highly conserved regions in the SSU rRNA alignment were used in the phylogenetic analysis.
A species phylogeny was created based on the NCBI taxonomic classification (http://itol.embl.de/other_trees.shtml). Since the relationships among some of the eubacteria lineages (Fig. 1) were unresolved, a SSU rRNA phylogeny was inferred using BayesPhylogenies (available from http://www.evolution.rdg.ac.uk/BayesPhy.html). This program implemented a joint model that accommodated shifts in site-specific substitution rates over time (ie, heterotachy) and among-site rate heterogeneity (ie, “pattern heterogeneity”)25,26; these phenomena are expected to occur in SSU rRNA-based phylogenetic analyses of prokaryote/eukaryote lineages. If heterotachy and “pattern heterogeneity” are not accounted for in phylogeny reconstruction, the resulting relationships are likely to be distorted. 26 A reversible-jump Markov chain Monte Carlo (rjMCMC) algorithm was used to determine how many distinct among-site rate-variation patterns, and branch length parameters (with a maximum of two parameters for each branch) were required to optimally describe the empirical data matrix. This approach is appealing because it requires far fewer parameters than conventional mixture models to describe heterotachy and “pattern heterogeneity”.25,26 A General Time Reversible (GTR) model of nucleotide substitution with discretized gamma-distributed rate variability (with 4 rate categories; Γ4) was employed. Three independent MCMC (Markov chain Monte Carlo) analyses (each with 3 chains running for 2 × 10 7 generations, sampling every 10 3 generations) were carried out to estimate the posterior distribution of phylogenetic trees, and post-burnin samples (with burnin set to 10%) from all analyses were combined. Convergence of the MCMC runs was determined by visually examining the cumulative posterior and between-run variation in split frequencies 27 using the on-line tool AWTY (“Are We There Yet”). 28 The software package FigTree (available from http://tree.bio.ed.ac.uk/software/figtree/) was used to generate the trees.
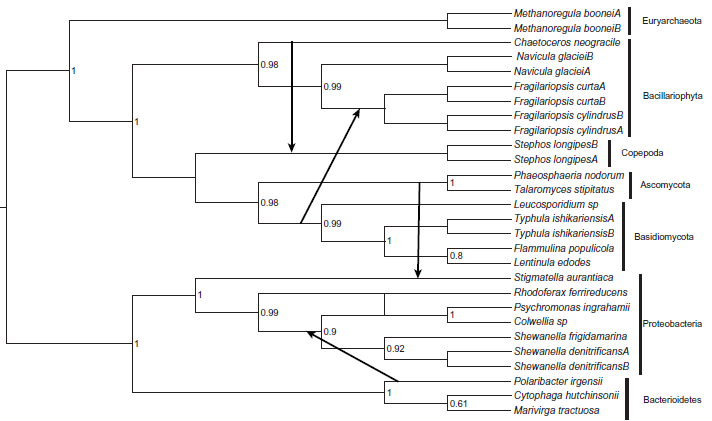
A rooted species phylogeny with the 4 horizontal gene transfer (HGT) events shown (see text for details).
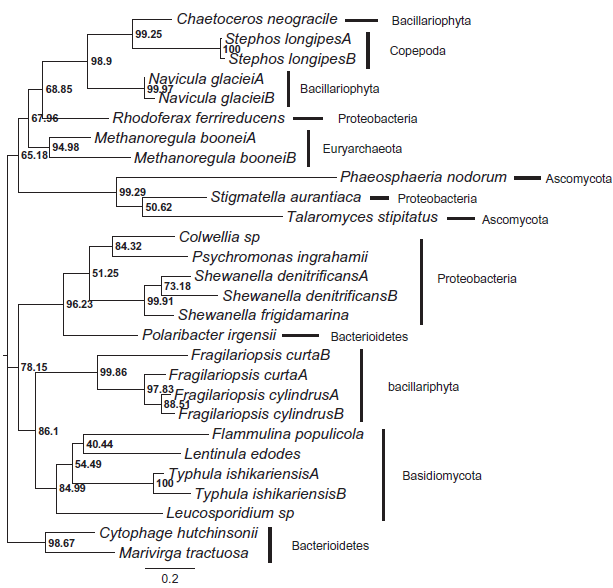
Unrooted maximum likelihood tree inferred by the Treefinder based on the alignment of the amino acid sequences.
The program Prunier (version 2.0) 18 which works in conjunction with Treefinder 29 was used to detect potential HGTs between the species included in this investigation. The “slow” Prunier method uses Kishino-Hasegawa, 30 Shimodaira-Hasegawa 31 expected likelihood weights 32 and the approximately unbiased 33 tests to infer whether topological differences between the gene and species trees are statistically significant. The “fast” Prunier method uses LR-ELW (Expected-Likelihood Weights applied to Local Rearrangements) edge support values 32 to identify such discrepancies. HGT events are inferred when statistically significant topological conflicts between the species and gene trees were identified. 18 Both the “fast” and “slow” methods, which in simulations have been shown to perform equally well 18 were used in the current study. The aligned amino acid sequences and the species phylogeny were provided as input for the Prunier runs. In each Prunier run, the gene trees were inferred by the Treefinder program. 29 For the “slow” method the default settings were used. The following settings were implemented for the “fast” method: boot.thresh.conflict = 95 (ie, support value threshold for topological conflict); fwd.depth = 1 (ie, maximal depth at which Prunier looks forward to find a significant HGT when the current HGT is not significant). The “boot.thresh.conflict” number is the minimum LR-ELW edge support value for a given node in the gene tree used for recognizing topological conflict between the gene and species trees. The “fwd.depth” (for further details see http://pbil.univ-lyon1.fr/software/prunier/) is the maximal depth at which the “fast” method Prunier looks forward to find a significant HGT when the current event is not significant. A depth value of 1 implies that if no significantly supported conflict can be removed with one HGT event the algorithm looks one step “forward” to see if the next HGT in the list will remove a significant conflict. If Prunier finds a better solution with a depth value of 2, it provides this solution in the output.
A newly developed unrestricted random effects branch-site model
19
was implemented for the purpose of detecting episodic diversifying selection on codons in the paralogs of the two
Results
Species and gene trees
The initial species phylogeny was generated based on the NCBI taxonomic classification information. This tree showed unresolved relationships for
Figure 2 shows the optimal unrooted maximum likelihood tree derived from the aligned amino acid sequences of the AFPs/AFLPs. A number of major conflicts are apparent between the species and gene trees (compare Fig.1 with Fig. 2). The prokaryotes
Horizontal gene transfer
The outcome of the Prunier analyses, based on the “slow” and the “fast” methods, were the same, namely the same HGT events were inferred (see Fig. 1). From here on the results of the “fast” method is described and discussed. The arrows in Figure 1 show the direction of HGTs. For these events to be inferred the topological conflicts between the gene and species phylogenies had to have a minimum LR-ELW edge support values of 95% in the former tree (Fig. 2). Thus, two transfers occurred among eukaryotes, that is from
Episodic diversifying selection analysis
Episodic evolution took place in 32% (15/47) of the branches in the

Relationships between the duplicated antifreeze protein genes in
Branches found to be under episodic diversifying selection by the unrestricted random effects branch-site model.

Branches (see Fig. 3 for branch numbers) that were inferred to be under episodic diversifying selection;
mean ω is the average dN/dS estimated for each branch under the free-ratio MG94 × REV model (no site-to-site rate variation);
ω–
q– values reflect the strength of negative selection (ω–) and the proportion of the total branch length affected by negative selection (q–);
ωN
qN -values reflect (nearly) neutral evolution (ωN) and the proportion of the total branch length affected by (nearly) neutral evolution (qN);
ω+
q+ -values reflect the strength of positive selection (ω+) and the proportion of the total branch length affected by positive selection (q+);
LRT is the likelihood ratio test statistic;
p is the uncorrected
corrected p is the probability based on Holm's multiple testing correction.
Discussion
Horizontal gene transfer
Conflicts between species and gene trees can be due to lateral gene transfers between distantly related species. 18 The program Prunier (see Material and Methods) has been demonstrated to perform well when it comes to identifying incongruences between species and gene trees caused by horizontal gene transfers. 18 This method allowed me to further investigate hypotheses about horizontal transfers of AFP and AFLP genes between distantly related taxa.
The Prunier analyses of the amino acid alignment rejected the idea that the prokaryote genera
Few studies, that provide evidence of eukaryotic-to-prokaryotic HGTs, have been reported.
13
However, the acquisition of an AFLP gene by the proteobacterium
One prokaryote-to-prokaryote transfer took place, that is between
Not all the proteins included here have been shown to have antifreeze properties (see Material and Methods). In database searches a number of genes show a high degree of similarity to those with known antifreeze functions—some of those sequences were included in the current investigation (also see discussion in
11
). Even though information about their distribution is scanty, many of the organisms that have AFLP genes are not known to occur in polar habitats nor in ice, suggesting that some of these proteins do not have a function related to cold tolerance. This is probably the case with
Episodic diversifying selection analysis
The results of the Prunier analysis suggested that the
Disclosures
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributorship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.
Footnotes
Acknowledegments
I thank James Raymond for introducing me to questions associated with horizontal transfer of antifreeze protein genes in diatoms and Vincent Daubin for advice on using Prunier. Three anonymous reviewers provided comments that improved the content of the paper.