
Abstract
Eukaryotic genomes are packaged into chromatin by histone proteins whose chemical modification can profoundly influence gene expression. The histone modifications often act in combinations, which exert different effects on gene expression. Although a number of experimental techniques and data analysis methods have been developed to study histone modifications, it is still very difficult to identify the relationships among histone modifications on a genome-wide scale.
We proposed a method to identify the combinatorial effects of histone modifications by association rule mining. The method first identified Functional Modification Transactions (FMTs) and then employed association rule mining algorithm and statistics methods to identify histone modification patterns. We applied the proposed methodology to Pokholok et al's data with eight sets of histone modifications and Kurdistani et al's data with eleven histone acetylation sites. Our method succeeds in revealing two different global views of histone modification landscapes on two datasets and identifying a number of modification patterns some of which are supported by previous studies.
We concentrate on combinatorial effects of histone modifications which significantly affect gene expression. Our method succeeds in identifying known interactions among histone modifications and uncovering many previously unknown patterns. After in-depth analysis of possible mechanism by which histone modification patterns can alter transcriptional states, we infer three possible modification pattern reading mechanism (‘redundant’, ‘trivial’, ‘dominative’). Our results demonstrate several histone modification patterns which show significant correspondence between yeast and human cells.
Background
Gene activities in eukaryotic cells are mainly regulated by transcription factors and chromatin structure. Chromatin fibers are composed of polymers of nucleosomes, which are the fundamental units of chromatin. Each nucleosome is composed of approximately 147 base pairs of DNA wrapped ~1.7 turns around a histone octamer consisting of two copies each of the core histones H2A, H2B, H3, and H4.1–3 On the one hand, the histones cover the DNA and prevent the genetic information from being accessed by many biological machineries, such as transcription, replication and recombination. On the other hand, the histones can be altered by the post-translational chemical modifications, including acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation, through which the interactions between histones and DNA can be altered.4,5 Among them, histone acetylation leads to a reduction of positive charges on the histone tails, and because of the negative charge of the DNA, less charge compensation leads to an open chromatin state, which is often associated with increased gene expression;6,7 histone methylation links to both gene activation and gene repression depending on its site and extent of methylation, and different effects may be associated with mono-, di-, or trimethylation of lysine residues. 8
A large number of residues within the histones are modified. In particular, a single modification can induce the occurrence of one or more subsequent modifications. Therefore, histone modification may form a ‘code’ and then comes histone code hypothesis. According to this hypothesis, covalent posttranslational modification of histone tails act sequentially or in combination to form a ‘histone code’ that is read by other proteins to bring about distinct downstream events. 9 However, this hypothesis has been much debated, some believed that use of histone modifications individually or sequentially cannot be considered a code since the total number of modifications do not necessarily contain more information than the sum of individual modifications; 7 others suggested that the existence of such a code should manifest itself such that different modification combinations lead to distinct outcomes.1,10 Although different interpretations of ‘histone code’ are possible, we believe that histone modifications function in a manner rated to their combinatorial effects rather than individual effect.
Many methods have been proposed to find the combinatorial effects. On the one hand, a number of experimental techniques have been developed to study histone modifications, such as, chromatin immunoprecipitation (ChIP), ChIP followed by amplification and microarray hybridization (ChIP-chip) and mass spectrometry (MS). On the other hand, some data analysis methods have been introduced to reveal the relationships between histone modification sites, such as, clustering methods11,12 and correlation analysis.13,14 But there are several disadvantages of the above methods. Specifically, clustering methods are good at clustering under all sites but difficult to cluster under subset of all sites, hence, they fail to identify whether the sites are functional or not. Meanwhile, correlation analysis can only reveal pairwise correlation between each pair of sites. In order to solve above problems, we proposed a method of association rule mining which not only overcomes aforesaid shortcomings but also successfully identifies the relationship among histone modification sites on a genome-wide scale.
Association rule mining was first proposed by Agrawal et al. 15 Also the Apriori algorithm was published by Agrawal and Srikant, and a method for generating association rules is described in this paper. 16 To improve the efficiency of Apriori, many variations of the Apriori algorithm have been proposed, such as application of hash tables to improve association mining efficiency studied by Park et al, 17 the sampling approach proposed in Hannu 18 and the partitioning technique discussed by Savasere et al. 19 Also, association rule mining has various expansions, such as cyclic association rules mining, 20 negative association rules mining 21 and market basket analysis. 22 Recent work23,24 has shown that the removal of hierarchically redundant rules from multi-level datasets by using a dataset's hierarchy is a promising approach to solving redundancy problem. To evaluate statistical significance of an association rules, chi-square test was used for statistical dependency between the antecedent and the consequent of the rule.25,26
This method is focused on combinatorial effects of histone modifications which have a significant effect on the gene expression. To identify these histone modifications, we have to rule out other factors which could have an impact on gene expression. Gene activities in eukaryotic cells are concertedly regulated by transcription factors and chromatin structure. Furthermore, since clusters of genes with the same histone modification patterns are enriched with coexpressed genes, the expression coherence (EC) scores are mainly contributed by transcription factors and histone modifications. As a result, if we eliminate the influence of TFs, we will gain transactions whose EC scores of their target genes are mainly attributed to same histone modification combinations. In this paper, through synthetically analysis of histone modification data, gene expression data, gene binding activity data and the relationships between transcription factors and chromatin modifiers, we identified Functional Modification Transactions (FMTs) and employed association rule mining algorithm and statistics methods to identify combinations of histone modifications. After applying this method to two distinct histone modification data, the specific modification combinations as well as global patterns of histone modifications were obtained.
Materials and Methods
Concept Descriptions and Notations
To better understand this method, we first present some useful concepts and notations used in the paper. First, we give some concept descriptions of transaction, candidate transactions, target genes of a transaction and EC score of a transaction. By definition, each transaction is a set of items (ie, itemset), while in this paper each transaction corresponds to a histone modification combination after discretization of histone modification data; candidate transactions are the specific transactions extracted from all transactions and prepared for data mining; target genes of a transaction is a cluster of genes with a common histone modification described by the transaction; similarly, EC score of a transaction means the EC score of the target genes.
Next, we give some notations used in this paper. For a transaction, each item is denoted as the form of ‘XY’, where X is the modification site of each gene,
Steps for the Method
Our method first identifies functional histone combinations according to gene expression and then applies association rule mining to candidate transactions. The functional histone modification combinations have two characters (1) significant EC scores (Materials and Methods) (2) no significant TFs in the promoter region or significant TFs which interact with chromatin modifiers. The steps of method can be described as follows: First, we generated 3
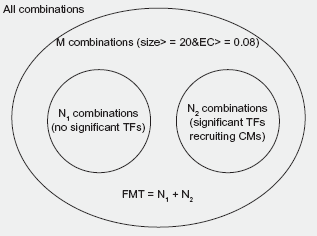
The Venn diagram of FMT.
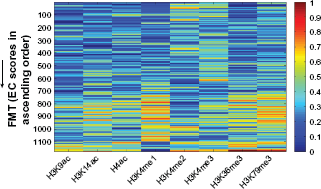
The global view of histone modification of FMTs for Pokholok et al's data.

The global view of histone modification of FMTs for Kurdistani et al's data.
Data Preparation
In this paper, datasets we utilized here mainly include histone modification, gene expression, and transcription factor binding activity data. The first histone modification data employed in this study is from Pokholok et al. 13 We chose 8 sets of histone modifications (H3K9ac, H3K14ac, H4ac, H3k4me1, H3k4me2, H3K4me3, H3K36me3, and H3K79me3), with H4ac referring to non-specific acetylation on any of the four sites, eg, H4K5, H4K8, H4K12, H4K16. The second histone acetylation data is originally from Kurdistani et al. 12 This data set includes measurements of the acetylation levels at 11 different sites (H4K8, H4K12, H4K16, H3K9, H3K14, H3K18, H3K23, H3K27, H2AK7, H2BK11, H2BK16). Since the acetylation levels of histones are affected by the occupancy of nucleosomes in that region, the acetylation data were divided by the average level of H3 and H2A histones from the Bernstein et al data set.27,28 The major difference between two data sets is that the acetylation in the first data set was measured against nucleosomal DNA while the second data set was measured against genomic DNA. In addition to above two histone modification datasets, expression dataset is necessary to evaluate the influence of modification patterns on expression. The mRNA expression profiles are combined by environmental stresses Gasch et al 29 and cell cycle Spellman et al 30 for 250 total conditions. To identify modification pattern from gene expression, we must eliminate the influence of TFs. Thus we extracted two disjoint subsets from all combinations of histone modification sites. One is the combinations whose promoters have no significant TFs identified by t-tests, of which the transcription factor binding activity dataset is from Young Lab Harbison et al; 31 the other is the combinations whose promoters only have TFs recruiting chromatin modifiers, among which the relations between transcription factors and chromatin modification factors are from Steinfeld et al. 32 Besides, the cell types and experiment conditions in various datasets are match or comparable (Table 1).
Histone Modification Data Discretization
To formulate histone modification profiles with transactions, we discretized histone modification sites of each gene. The value greater than 60th percentile, less than 40th percentile were regarded as ‘over-expressed’, ‘under-expressed’ respectively. To avoid ambiguity due to measurement noise, the middle 20% of genes was regarded as ‘NULL’. 33
Summary of various datasets.

Expression Coherence Score of a Transaction
In order to determine whether the target genes of a transaction were significantly correlated in their mRNA expression profiles, we calculated all pair-wise Pearson correlation coefficients for the expression profiles of all the genes within a transaction. For a transaction with
Identifying the Threshold of EC Scores
We first randomly selected 100,000 samples with the size of 20, and selected a threshold (0.08) so that only 5% (95% confidence) of the random samples pass this threshold. Then we iteratively performed above process with different sample sizes, such as 30, 40, 50, etc., and found that the larger the size of sample, the smaller the threshold is (data not shown). Therefore, we identified the strictest threshold with the sample size of 20, and considered the transaction was significant if it's EC score was above this threshold (Fig. S2).
Identifying the Histone Modification Regions of Different Sites
Since we obtain histone modification patterns according to gene expression (EC score), only the patterns that make a significant contribution to EC score can be found. Therefore, the choices of regions of different sites are based on the contribution of each site to gene expression respectively. Pokholok et al's histone modification data given in genome-wide location format, was separated into there regions, TSS, ORF and promoter. According to genome-wide distribution pattern of histone modifications, 34 acetylation level of H3 and H4 as well as H3K4me3 peak close to TSS region, hence, we assign the mean modification level of TSS region to these sites. Likewise, the remaining sites peak at ORF region, so we assign the mean modification level of ORF region to remaining sites. Moreover, correlations between the mean region modification levels and transcriptional activity are mostly larger than other regions. Besides, transcriptional activities correlate highly with EC scores (r = 0.53, for 1183 FMTs). Taken together, the regions we selected have a significant contribution to EC score. For Kurdistani et al's data, we used acetylation data from intergenic regions for two reasons. First, the data was already separated into intergenic region and ORF region according to each gene. Second, histone acetylation correlate positively with transcription levels and highly enriched in promoter regions. 3
Identifying Functional Modification Transactions (FMT)
As we know, gene expression is mainly regulated by transcription factors and histone modifications. In order to identify combinatorial effects of histone modifications, we should extract transactions whose expressions of their target genes are contributed by their corresponding histone modifications, or rather, these histone modifications are functional, and the extracted transactions are defined as Functional Modification Transactions (FMT). We carried out following steps to select FMTs (Fig. 1). First, for 3
Basic Concepts of Association Rules
Association rules discovery technique finds interesting associations or correlation relationships among a large set of data items. This method extracts sets of items that frequently occur together in the same transaction, and then formulate rules that characterize these relationships.
Let 
We take an example to illustrate above concepts. For Pokholok et al's data,
Rules extracted from Pokholok et al's data.

By the way, we call the confidence of a rule with empty antecedent the priori confidence, and the confidence of a rule with non-empty antecedent we call the posterior confidence. Generally, rules that satisfy both a minimum support threshold and a minimum confidence threshold are called strong. In data mining, associations can be generated using this “support-confidence” framework, but not all of rules satisfying the minimum support and minimum confidence thresholds are interesting to the user. Let's examine the simple example and let us assume that 60% of all customers buy some kind of bread, ie, the rule ‘⇒
Statistical Significance of Association Rules
The above measures do not involve information about statistical significance. The chi-square measure is often used to measure the difference between a supposed independent distribution of two discrete variables and the actual joint distribution in order to determine how strongly two variables depend on each other. A
Since it is possible to find a biological function associated with a histone modification, we carried out a similar analysis on 1000 randomly generated candidate transaction sets to test the novelty of the combinations detected by our method. As a result, the ratios of explicable rules for these randomized data are much smaller than that for the real data (data not shown). Therefore, the association rules which can be explained by biological function are significant, and then the detected combinations supported by biological function are novel.
Elimination of Redundant Association Rules
From the rules extracted from transactions, we found some redundant rules, such as, 13, 63, 81 ⇒ 51 and 13, 63, 81, 71 ⇒ 51. Because the association rules containing redundancies are difficult to comprehend, we sought methods to deal with this issue of redundancy. According to definition below by Gavin Shaw we can remove hierarchically redundant rules.
Definition (Hierarchical Redundancy for Approximate Basis): Suppose that two approximate association rules,
Specifically, we take an example to manifest how to eliminate redundant rules. For rule1 (
Results
In this paper we propose a method to identify the combinatorial effect of histone modification by association rule mining. The technology of association rule mining can find interesting association or correlation relationships among a large set of data items. We adopt this data mining technology to explore the relationships or the synergies between these histone modifications. In short, this method first identifies functional histone combinations according to gene expression and then extracts correlation relationships or patterns among these large histone combinations by association rule mining technology. In the following sections, we describe extracted rules and patter ns from Pokholok et al's data and Kurdistani et al's data.
Association Rules Derived from Pokholok et al's Data
We first applied our method to Pokholok et al's data and obtained some intriguing relationships. Specifically, we selected acetylation levels at 3 sites, H3K9, H3K14, and H4 (referring to non-specific acetylation on any of the four acetylable lysines on H4 tails), and 5 methylation levels of H3K4me1, H3K4me2, H3K4me3, H3K36me3, and H3K79me3. The above eight histone modification levels are measured in YPD medium. According to genome-wide distribution patterns of histone modifications,
34
we selected the histone modification levels near TSS (transcription start sites) for H3K9ac, H3K14ac, H4ac, and H3k4me3, and selected the mean level of the ORF for the rest modifications(Materials and Methods). As shown in Fig. S1, among 3
8
transactions, we filtered out transactions whose number of target genes is less than 20. Then we got 3052 transactions, and after being filtered by EC scores, 0.08(identifying the threshold of EC scores, Fig. S2), 1674 transactions were obtained. We obtained 552 N1 transactions and 631
Overview of the Extracted Rules
We can see the global view of histone modification of FMTs (Fig. 2). In Fig. 2, the higher the EC scores, the higher the histone modification level is, which confirmed the FMTs we chose were reasonable. From the overall over-expressed state of extracted 21 rules (Fig. 4), we found that a majority of rules (61.9%, 13 of 21) includes item ‘63’ (the modification level of H3K4me3 is over-expressed). It is consistent with the fact that hisone H3K4 methylation is a post-translational modification that is exclusively associated with actively transcribed genes.37,38 There are 71% (15 of 21) of rules each of which is consisted of any of ‘63’ (H3K4me3), ‘73’ (H3K36me3), or ‘83’ (H3K79me3), and synergized with ‘13’ (H3K9ac), ‘23’ (H3K14ac), or ‘33’ (H4ac), such as 81, 13, 63 ⇒ 51, 41, 33, 73, 83 ⇒ 53, etc. These results indicate that actively transcribed euchromatin has high level of acetylation and is trimethylated at H3K4, H3K36 and H3K79. 5 There are 90% (19 of 21) of rules each of which has an over-expressed histone modification level at sites within H3K9ac, H3K14ac, H4ac, H3K4me2, and H3K4me3. Out of 13 rules which includes ‘63’ item, there are 10 rules(account for 77%) that emerge with ‘13’ or ‘23’, which suggest that H3K4me3 and H3 hyperacetylation occur together. 39 Yet, intriguingly, there are 52% (11 of 21) of rules can be explained by the previous literature. In the following sections, we discuss the patterns extracted from Pokholok et al's data.

Fraction of over-expressed or under-expressed state at sites from extracted 21 rules for Pokholok et al's data.
Cross-Talk between (H3K9ac|H3K14ac|H4ac), H3K4me2, H3K4me3 and H3K79me3
From rule1 to rule4 ([81, 13, 63 ⇒ 51], [81, 13, 51 ⇒ 63], [81, 23, 51 ⇒ 63], [81, 33, 51 ⇒ 63], see Table 2), we found that there was a similar pattern among them, which can be formulated as H3K9ac [+] or H3K14ac [+] or H4ac [+], H3K4me2 [–], H3K4me3 [+] and H3K79 [–]. Since association rules were extracted from FMTs whose target genes have higher EC scores, the above synergic patterns are indispensable to active genes. During gene activation state, TFs at the upstream activator sites recruit positive modifiers, such as histone acetylases (HAT), at the promoter, while DNA-bound RNA polymerase recruits histone methylases at the ORF. 2 In addition, previous studies also showed a correlation between H3K4me3 and RNA polymerase (POL) II initiation. 40 Moreover, acetylation of H3 and H4 or dior trimethylation (me) of H3K4 are associated with active transcription. 34 Taken together, it is reasonable that the sites of H3 or H4 acetylation and sites of H3K4 methylation coexisted in this pattern, such as H3K9ac, H3K14ac, H4ac and H3K4me2, H3K4me3.
We also found interesting phenomena in above rules. First, H3K79m3 are under-expressed in the above pattern. Since little is known about the function of H3K79me3, the mechanism under this pattern is still unknown. Next we observed that the level of H3K4me3 is over-expressed while H3K4me2 is under-expressed. It is unclear, however, what reasons lead to this exclusive relationship between H3K4m2 and H3K4me3. We speculated that there may be two possibilities behind this phenomenon. First, like their roles in transcription, H3K4me2 is related with potential for gene activity, whereas H3K4me3 is related with gene activity. Second, it is also possible that H3K4me2 and H3K4me3 are at different methylation state, 34 therefore, there are exclusive at active genes. Then we also found that H3K4me3 always was associated with H3 acetylation. The reasons are described as follows: One the one hand, because Gcn5 HAT complex stimulates H3K4me3, H3 acetylation are associated with H3K4 metylation; on the other hand, Chd1 chromodomain links H3K4 methylation with H3 acetylation. 39 The above evidence highlights the interplay that can occur between acetylation of H3 or H4, H3K4me2, H3K4me3 and H3K79me3.
Cross-Talk between (H3K4me2|H3K4me3) and (H3K9ac|H3K14ac|H4ac)
This pattern is more universal than above pattern, there are 9 rules (rule1–4, rule9–12, rule15) which satisfy this pattern. In this pattern, H3K36me2 and H3K36me3 are mutually exclusive, while H3K9ac and H3K14ac and H4ac are exclusive with each other. As described above, both H3K4me2 and H3K4me3 are enriched at actively transcribed genes, which is supported by analyses of the different H3K4 methylation states and their distribution in various organisms. 8 Furthermore, H3K4me3 always promote H3 or H4 acetylation. Taken together, this pattern is reasonable and easy to interpret.
Interestingly, this pattern is highly consistent with notable patterns of coexisting histone marks in human cells, such as ‘H3K4me2/3 + H4K16ac’ and ‘H3K4me2/3 + H3K9/14/18/23ac’, which were supported by chromatin immunoprecipitatation (ChIP) and mass spectrometry (MS) method. 41 Specifically, from the pattern (H3K4me2|H3K4me3) + (H3K9ac|H3K14ac|H4ac) in yeast, we can see that H3K4me2 or H3K4me3 cooperate with H3K9ac or H3K14ac or H4K5acK8acK12acK16ac (H4ac referring to non-specific acetylation on any of the four sites, eg, H4K5, H4K8, H4K12, H4K16). The above pattern is highly consistent with pattern ‘H3K4me2/3 + H4K16ac’ in human cells, and apart from H3K18ac and H3K23ac sites, the other pattern ‘H3K4me2/3 + H3K9/14/18/23ac’ in human cells is also consistent with that yeast pattern. In general, the above yeast pattern nearly corresponds to combination of two patterns of human cells. Therefore, we deduced that there are significant correspondence between yeast pattern and pattern of human cells.
There are several similar histone modification characteristics between human cells and budding yeast, such as, association between H3K4me3 and RNA POL II initiation, association between H3K36me3 and elongation, association between H3K4 methylation and actively transcribed genes, etc.3,40 Therefore, one possible interpretation is that this pattern can be applicable to human cells. Nevertheless, further experimental methods need to be done to validate this pattern.
Cross-Talk between (H3K14ac|H4ac), H3K4me2, H3K36me3 and H3K79me3
We also observed similar pattern form rule16 (41, 33, 73, 83 ⇒ 53) and rule21 (41, 23, 73, 53 ⇒ 83). This pattern is consistent with activation process of gene expression. During activation state, DNA-bound activator firstly recruits histone acetylases (HAT) at the promoter, whereas DNA-bound RNA POL recruits histone methylases at the ORF. Secondly, early in elongation, phosphorylation of C-terminal domain (CTD) polymerase result in recruiting COMPASS complex, part of which (Set1) methylates H3K4. Last, later during elongation, phosphorylation of the CTD results in recruiting Set2 methyltransferase which methylates H3K36. 2 Besides, H3K36m3 and H3K79me3 are implicated in activation of transcription, and histone H3K36 methylation mediated by Set2 is an important landmark on chromatin during elongation.5,34 Thus, it is reasonable in above extracted pattern. Since little is known about the function of metylation of H3K79, the exact mechanism by which H3K36me3 cooperates with H3K79me3 remains to be determined.
Association Rules Derived from Kurdistani et al's Data
In addition to Pokholok et al's data, we also tested our method on Kurdistani et al's data, and several obtained relationships are validated. In this dataset there are 11 acetylation sites, including three lysines in histone H4 (K8, K12, K16), four in histone H3 (K9, K14, K18, K23), one in H2A (K7), and two in histone H2B (K11, K16). Here, we utilized acetylation data for intergenic regions (Materials and Methods). Among 3
11
transactions, we filtered out transactions whose number of target genes is less than 20. Then we got 10941 transactions, and after filtered by EC scores (0.08), 2193 transactions were obtained. We obtained 594
Rules extracted from Kurdistani et al's data.

Overview of the Extracted Rules
As shown in Figure 3, we found that ‘63’ (H3K18) is the largest abundance with the ratio of 71% (968 of 1354 FMTs). Furthermore, H3K18 was identified as the most widely regulated acetylation site, and its acetylation appears to be a general mark of active transcription,12,27 thus we deem that H3K18 perform its function in a global manner. On the other hand, from the overall under-expressed state of extracted 69 rules (Fig. 5, after elimination of H3K18), we found that H4K16 accounts for the largest proportion among extracted rules. The reason may be that the number of TFs to regulate H4K16 is the least among 11 acetylation sites, which is only 2 TFs to regulate H4K16 contrast to 15 TFs for H3K18. 27
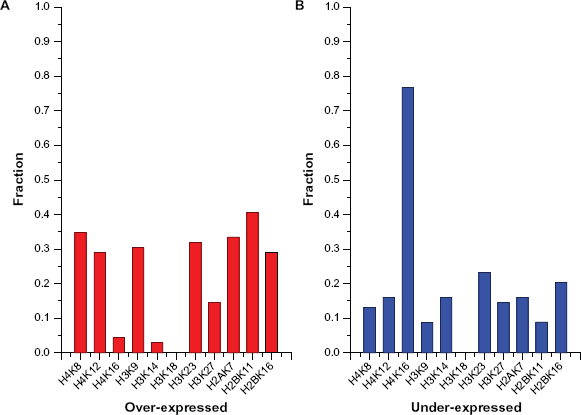
Fraction of over-expressed or under-expressed state at sites from extracted 69 rules for Kurdistani et al's data.
Cross-Talk Involving Unacetylation of H4K16 and Acetylation at other Sites
From Table 3, we found that there was a common item (31) from a large majority of extracted rules. Therefore, the item of ‘31’, ie, unacetylation of H4K16, is important for these histone modifications and plays a special role in transcription. Furthermore, the unacetylated H4K16 has an activating role in euchromatin and unacetylated H4K16 in the context of acetylation at other sites is important for transcription. 6 For example, among 32 extracted rules (supports are greater than or equal to 1, see Table 3), there are 27 rules which are consistent with role of unacetylation of H4K16 in the context of acetylation at other sites, such as, H4K8, H3K23, H3K9 and so forth.
Discussion
We have proposed a new method to identify the combinatorial effects of histone modification by association rule mining. The method is based on association rules discovery technique and can find individual functional sites as well as the combinations of them. Through the analysis of two histone modification datasets, we extracted a number of rules some of which are strongly supported by previous studies. The drawback of association rule mining is that the number of generated rules is often very large, even if minimum support and confidence are satisfied. To address this question, besides support and confidence adopted, we also adopted following methods: First, we restricted association rule with a single item in the consequent. Second, a high improvement cutoff (is larger than 110%) must be satisfied for each rule, which guarantees strong dependence between antecedent and consequent (Materials and Methods). Last, the other two measures,
The study obtained two sets of association rules and patterns from two different datasets with little overlap. We consider that the selection of modification sites is the major cause of this non-overlapping. Specifically, for Pokholok et al's data, there are eight sets of histone modifications (H3K9ac, H3K14ac, H4ac, H3K4me1, H3K4me2, H3K4me3, H3K36me3, and H3K79me3) whereas for Kurdistani et al's data, there are 11 acetylation sites (H4K[8,12,16], H3K [9,14,18,23,27], H2AK7, H2BK [11,16]) without methylation sites. Strictly speaking, there are only two common sites (H3K9ac, H3k14ac) between two datasets (H4ac denotes H4K5ac8ac12ac16ac). In addition to addressing the above questions implicated in this method, we sought to understand how these patterns could influence gene expression, and the mechanisms underlying these combinatorial effects. To our knowledge, histone modification can change transcriptional states through two mechanisms.
42
The first mechanism implicates recruitment of proteins or complexes through binding of their specific protein domains to acetylated or methylated hisones. For example, the bromodomain recognizes acetylated lysine residue whereas chromodomain recognizes methylated lysine residue, which are found in many chromatin-regulating proteins. The second mechanism involves the formation of a less compact nucleosome structure. Because positively charged histone tail interacts with the negatively charged DNA in a nucleosome, acetylation neutralizing the positive charge of the lysine results in a destabilized nuelosome structure. For Pokholok et al's data, we extracted 777 genes from 21 rules, while 857 genes from 32 rules from Kurdistani et al's data. We observed very little overlap (174 genes, hypergeometric
In addition, the genes (1460 genes) mentioned above contain a high percentage of genes (hypergeometric
Interestingly, there are also a few novel rules which contradict with extracted patterns. For example, rule5 (13, 71, 33, 63 ⇒ 51) and rule6 (13, 71, 23, 63 ⇒ 51) have either H3K9ac and H4ac or H3K9ac and H3K14ac in the antecedent, which contradict with the above statement that these acetylation sites are exclusive for each other. One possible explanation could be that they are mutually exclusive in some patterns, but not in others; another possible explanation is that they are exclusive, and the contradiction is due to noise. Which statement is correct remains to be elucidated. Interestingly, there exist common modification patterns between yeast and human cells, which could guide further experimental methods to explore histone modification patterns. Besides Chip-chip, Chip-SAGE and Chip-Seq experimental approaches, knockout experiment is a good choice to study modification patterns.
Conclusions
We have developed a method for the analysis of histone modification data, which can identify the combinatorial effects of histone modifications. This approach is based on the association rules discovery technique and statistical hypothesis testing method. We applied this algorithm to two different datasets and generated many interesting rules and patterns. From Pokholok et al's data, we acquired 21 rules. From the landscape of extracted rules, we found the dominant signal of H3Kme3, which could be supported by many literatures. Also, we extracted three typical patterns, such as, ‘(H3K9ac|H3K14ac|H4ac) + H3K4me2 + H3K4me3 + H3K79me3’, ‘(H3K4me2|H3K4me3) + (H3K9ac|H3K14ac|H4ac)’, and ‘(H3K14ac|H4ac), H3K4me2 + H3K36me3 + H3K79me3’, which also can be explained by previous studies. Most interestingly, our results demonstrated a very good correspondence between yeast and human cells regarding the dominant role of H3K4me3 and the pattern ‘(H3K4me2|H3K4me3) + (H3K9ac|H3K14ac|H4ac)’. We also obtained several global factors from Kurdistani et al's data. Because over-expressed state of H3K18 occurs in large majority of rules and its acetylation appears to be a general mark of active transcription, we deem that H3K18 perform its function in a global manner. Due to the global role of H3K18, we eliminated rules which contained ‘6X’ (X = 1 or 3) to highlight histone modification patterns of other modification sites. After elimination of H3K18, we found synergy between unacetylation of H4K16 and acetylation at other sites, which also be supported by literatures. Furthermore, these novel patterns we extracted lay a useful foundation for the additional experiments necessary to gain a fuller understanding of the roles of combinations of histone modifications in gene expression. More importantly, the method used here to identify the combinatorial effects of histone modifications can also be used to gain insights into relationships between histone modification sites across the genome in other higher eukaryotes.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgements
JW and XD designed the study, analyzed the results and drafted the manuscript, and JW also implemented the algorithms, carried out the experiments. XQ, YD, JF, ZD and CH participated in the analysis and discussion.