
Abstract
RNA molecules have been discovered playing crucial roles in numerous biological and medical procedures and processes. RNA structures determination have become a major problem in the biology context. Recently, computer scientists have empowered the biologists with RNA secondary structures that ease an understanding of the RNA functions and roles. Detecting RNA secondary structure is an NP-hard problem, especially in pseudoknotted RNA structures. The detection process is also time-consuming; as a result, an alternative approach such as using parallel architectures is a desirable option. The main goal in this paper is to do an intensive investigation of parallel methods used in the literature to solve the demanding issues, related to the RNA secondary structure prediction methods. Then, we introduce a new taxonomy for the parallel RNA folding methods. Based on this proposed taxonomy, a systematic and scientific comparison is performed among these existing methods.
Introduction
Bioinformatics is a new discipline resulting from the combination of two science fields: Computer Science, and Biology. This new scientific application has grown rapidly and nowadays it is becoming a cornerstone for each molecular biological study.
1
It utilizes computer implementations and algorithms for collecting, accumulating, storing, analyzing, and integrating biological data and genetic macromolecules like deoxyribonucleic acid (DNA), ribonucleic acids (RNA), or proteins. The DNA has directions on how to build other components of cells, such as proteins and RNA molecules. The RNA is a type of nucleic acid that provides a mechanism to copy the genetic information of the DNA. There are two different types of RNA which are the Messenger RNA (mRNA) and the Transfer RNA (tRNA). They play important roles in the living cells and protein synthesis. Recently, researchers have found that they can use the RNA interference (RNA
It is important to study the folding of RNA molecules to understand their roles and functionalities. 6 Medical researchers and biologists can find different vital roles of RNA molecules by scrutinizing their RNA secondary structures. This will pave the way in front of biomedical researchers to utilize the RNA molecules in useful application like when there are used for productive treatments. 4 The experimental methods that are commonly and broadly used for determining three-dimensional (3D) structures of RNA are listed as follows: X-ray crystallography and Nuclear Magnetic Resonance (NMR). In a biological context, these experimental methods are the most prominent accurate methods to determine the RNA tertiary structure, which is a 3D structure. But, both of these physical methods are time-consuming, expensive and computationally difficult to accomplish. To give the reasons, which make these purification techniques for determining the RNA 3D structures are tedious and difficult. In the rest of this section, we elaborated more about how both of these experimental methods are carried out by chemical or biological researchers.

Due to these constraints and difficulties from the experimental physical sides, computational methods from computer scientists and bioinformatic researchers have become more demanding to do the RNA structure prediction process. Predicting the RNA 3D structure from the primary sequence is difficult to accomplish. Hence, the bioinformatic researchers first detect and predict the RNA secondary structure from a given RNA primary sequence. Then, the result of this RNA secondary prediction process will assist the biologists to determine the RNA tertiary structure.
Thus, solving the RNA secondary structure problem is becoming a main issue among bioinformatic researchers. 10 Recent approaches and current work concentrate on applying the parallel techniques to the previous RNA computational algorithms. In this paper, the works of the researchers explore the state-of-the-art of parallel techniques were proposed for solving RNA structural problems.
The earliest prediction methods for solving the RNA secondary structure problems were presented by Waterman and Smith
11
and Nussinov et al.
12
These computational algorithms proposed two different methods for the RNA/DNA folding structure, which require
Basically, most computational algorithms, approaches and methods for solving the RNA secondary structures problems were restricted to the length of the bases, which were only a few hundreds of characters. Computational methods become much more desirable to solve traditional RNA cases and original RNA algorithms. 15 Then subsequently, promising passageway simplifies these constraints and solves the bottleneck for the original RNA computational prediction algorithms, by implementing them on new parallelization approaches. Some of the more parallel popular approaches to reach this objective are to design them on the Field Programmable Gate-Array (FPGA), the Graphics Processing Unit (GPU), the multi-core or the cluster master-slave parallel architecture systems. 16
Schematic Study
We begin this comparative taxonomy study for the RNA parallel prediction methods by compromising and organizing this paper into three main research issues; (i) literature and background of the RNA predictions, (ii) RNA research models, (iii) the discussion and evaluation of existing methods with a comparative taxonomy. The main objective of this research is to present a comprehensive summary of the state-of-the-art researches on the RNA secondary structure predictions methods. Thus, we combined and joined among these all parts as follows:
Finally, there is the discussion and the evaluation of existing methods with a comparative taxonomy. The researchers proposed and described a general taxonomy for RNA parallel methods. By using this taxonomy, they applied a comparative process for the previous existing parallel RNA prediction methods. This process enabled the researchers in this field to distinguish among the parallel RNA prediction approaches. Future researchers will be able to choose the suitable parallel paradigm that will fit their interests. This selection will depend on the group of the RNA molecules that the bioinformatic researchers working on or needs.
Background
The RNA molecules had been confirmed to be very resourceful materials by.3,4 The RNA molecules play different crucial functions and roles in living organisms and in many biological processes. Recently, it became clear that the RNA molecules play various roles, not just an intermediate in protein synthesis. But also, the latest researches are looking to utilize the RNA
RNA chemical structure
To understand the roles and functions of the RNA molecules, the biomedical engineers and researchers need to determine and scrutinize the RNA tertiary structures. As the first step to reach this goal, they ought to know the RNA molecules chemical structure and the motives that forced them to make these folds of 3D structures. Accordingly, the RNA was the common name for ribonucleic acids; they are made of long chains (single-stranded) of nucleotide units. There are three different components of the RNA nucleotides: the nitrogenous base, the sugar, and the phosphate group. While, the RNA backbone is made up of ribose five atom carbon-sugar counted from 1′ through 5′ and it is attached by two phosphate groups in 3′ and 5′, respectively. From the sugar group “ribose”, the RNA molecule acquired its nickname “Ribonucleic Acids”. The nitrogenous bases in the RNA group were made up of four different bases Adenine (A), Cytosine (C), Guanine (G), and Uracil (U). These bases attached to the five-carbon sugar in 1′ position and they give RNA molecules characteristic possessions and properties.
Veritably, the phosphate groups in the backbone of the RNA have a negative charge, which makes the RNA a charged molecule. 9 Due to this, the charged RNA molecule inside the living cells is not stable. Thus to gain more stability, some parts of the single-stranded RNA fold back on itself forming double helices. This RNA folding process makes the determination methods intricate and not easy to determine the RNA 3D structures. The details about the RNA molecules and its chemical structure was explained in. 22
The RNA primary and secondary structure
Both the phosphate groups are attached to the 3′ and 5′ positions from ribose sugar in the backbone of RNA molecules. Due to this fact and based on the convention and general agreement among biologists, the RNA primary structure is a string series of bases reported from the 5′ end to the 3′ end, as shown in Figure 3(a).

The main systematic chain steps of RNA research study.

The RNA secondary structure derives from the pairing up of these four nucleotides according to the rules of
Problem Statement
Recently, RNA molecules have been confirmed to be very resourceful materials in the medical process and biological systems. 23 Biologists can determine the RNA tertiary structure from its secondary structure. The biologists need this 3D structure of the RNA molecule to derive its function and essential role. Thus, the bioinformatic researchers introduced computational prediction methods, which can predict the RNA secondary structures from a given RNA primary sequence. In Figure 2, the authors depictured and explained the vital crucial position of the RNA secondary structure in the chain of the RNA research. Also, this figure showed the flow chain of the RNA research study, consecutively. Moreover, it confirmed that the computational secondary structure prediction processes is a required preliminarily step for determining the RNA 3D structure.
The RNA folding recognition methods attempt to predict an accurate and more stable RNA folding structure based on the Minimum Free Energy (MFE) models. As shown in Figure 3 (d), the nature of some types of the RNA structure forms the pseudoknots, in some parts. The RNA molecules with the pseudoknots structure make the calculation process of the RNA secondary structure prediction algorithms complex. These complexities of the RNA prediction algorithms conform and confirm the execution time and memory storage space, computationally.
The components of the RNA involved in understanding the RNA functions, which are extracted from,
24
and can be presented as follows:
RNA primary sequence structure is a string of A single-stranded RNA secondary structure is a list of base-pairs which can be viewed as a set of, X, forms on acceptable base pairs ( These “ In addition, these base pairs ( It should be at For all base pairs ( Thus, namely two bases that form the canonical pair must be located at different locations. While, the RNA sequence does not fold back on itself too sharply. Also, each base can be paired and combined at most only with another base. Restrictedly, the implement-ers allowed just “
Thermodynamic algorithm for RNA prediction
From the energy stability point of view, the phosphate groups in the backbone of RNA molecule have a negative charge. Thus, the RNA molecules inside the living cells are not stable.9,27 They will fold back on themselves to reach more stability.
Then, the main goal of the RNA secondary structure prediction computational method is to arrive at more stable equilibrium of the RNA folding form, based on the free-energy model. Hence, to calculate the RNA free energy stabilities, it is necessary to predict RNA secondary structure by calculating the MFE, which is named as an optimal RNA structure. 28
The summation of energies for all loops is the energy of RNA secondary structure (Eq. 1), which extracted with condensation from.18,28,29 The empirical calculations explained and confirmed that over 99% from the execution time for the RNA prediction algorithm is in computing the MFE. 20

In reality, to the best knowledge of the authors, thermodynamic prediction approach for calculating the energy of the optimization RNA, had been expressed and introduced for the most time by Lyngso
These MFE calculative motifs complicate the general pseudoknotted RNA secondary structure prediction algorithms. The pseudoknots type turns the RNA prediction algorithms to be
Consequently, researchers of pseudoknotted RNAs faced with three problems:
Existing Sequential Methods
Basically, one of the important tasks in front of bioinformatics and computer application researchers is the RNA secondary structure prediction dilemma. From a biological point of view, there are various complexities and constraints that are faced the biomedical researchers in experimental methods to determine the RNA tertiary structures.
7
Recently, many computational efforts have been presented on the computer side, to detect the RNA secondary structure from the primary sequence. The predictive approaches have been introduced to solve the related issues in the RNA structural detection field as follows: (
The original existing methods that have been proposed in solving the RNA secondary structures are divided into two major groups. The first group predicts the
Then, the researchers in this paper classified these sequential RNA prediction methods in a schematic diagram, as shown in Figure 4. Next, the researchers converged these RNA prediction methods, as shown in Table 1, which was adapted and extracted from. 22 This table included the most well-known existing RNA sequential prediction methods and approaches that have been produced lately to predict RNA secondary structures.

Schematic diagram of RNA structural prediction methods.
Existing sequential methods for RNA secondary structure prediction.

Parallel Methods and Schemes
The experimental methods are completely accurate for determining the RNA 3D folding structures. But due to their time consuming and expensive nature, many computational approaches have been proposed to predict the RNA secondary structure, which includes: (
Lately, many different parallel methods were introduced, in order to face the computational complexities of the RNA secondary structure prediction problem such as: (
Multi-core RNA parallel algorithm
Recently, the mainly accepted and accurate approaches for predicting the RNA secondary structures sequentially are
In fact, the value and the significance of the
Therefore, the
Parallel algorithm on GPU
The Graphics Processing Units (
Basically, the researchers utilized the latest modern GPUs to speed-up the algorithms in solving RNA secondary structure problems. Rizk and Lavenier in
16
explored a new implementation for the previous function, which was used in solving RNA prediction problems. The researchers re-implemented the
The significance of the research results in, 16 is to obtain faster execution time in the RNA secondary prediction algorithm. Utilizing the GPU design, reduced the execution time and the computation complexities without any extra cost. This improves the results when compared with the other competitive RNA prediction methods like the Multi-core, the clusters or the multiprocessors systems.
Parallel framework on cluster
The computer hardware Beowulf cluster showed some strong parallel features, based on the master-slave paradigm. 49 Many researchers exploited this parallel architecture to compute the traditional RNA secondary structure detection methods. The parallel implementation on the Beowulf cluster was utilized for re-implementing the original RNA prediction methods. 17 This work pointed out that there were good results with a higher accuracy and a faster execution time in the RNA structural algorithms, comparing to the original RNA detection methods.25,34 The pseudoknotted RNA complicated structure algorithms could also benefit from this parallel design. Despite those RNA pseudoknots structures were always recognized in most of the RNA molecules and they were known among RNA researchers. 24 But, due to that the pseudoknotted RNA molecules are computationally demanded nature. Thus, the RNA pseudoknots types mostly ignored from many RNA prediction methods. Namely, some researchers compute their RNA prediction methods without pseudoknots to get more simplicity. 34 These perdition methods to solve RNA secondary structures would be inaccurate when they neglected pseudoknotted types.
A prominent work has opened a new epoch in pseudoknotted RNA secondary structure prediction research. The
The main contribution of the
Parallel on FPGA co-processors
Different researches found and confirmed that the traditional RNA prediction methods can be put into operation by using fine-grained hardware implemented on the FPGA. Recently, the bioinformatic researchers found that the modern computers, parallel or multi-core, do not show a greater than 50% parallel usefulness and efficiency. 18 But the researchers achieved better accelerations when using the FPGA co-processors, which grows to be a hopeful approach for the main RNA prediction algorithms like RNAalifold, 18 Nussinov's algorithm 19 and Zuker's algorithm. 20
(i) Fine-grained parallel implementation on the FPGA for the RNAalifold method
The most popular computational approach for RNA secondary structures based on using the MFE-model is the
The researchers in
18
supplied an innovative accelerated approach for the
(ii) Fine-grained parallelization of Nussinov's algorithm implemented on FPGA
Many RNA folding methods utilized empirical models for predicting the RNA secondary structures based on the MFE estimation by using the Dynamic Programming (DP) algorithm. Jacob et al in
19
pointed out that in all these empirical models, algorithms for predicting RNA structures are just DP recurrences from the original Nussinov's algorithm.
12
There are two features that make this algorithm computationally more applicable for predicting RNA secondary structures among the other prediction methods; Firstly, Nussinov's algorithm used the length of RNA sequence as a proxy for the MFE and it computed the most maximum of RNA base pairs. Secondly, the original Nussinov
12
one runs in
Jacob et al in
19
adapted one of the most recent parallel FPGA architectures (
(iii) Parallel fine-grained implementation for the Zuker algorithm on FPGA
The most well-liked and admired computational approach for the RNA secondary structure based on using the MFE is the Zuker's algorithm.
13
It was confirmed to be the most stable RNA secondary structure prediction method based on calculating MFE. The Zuker's algorithm runs in
The promising way to make Zuker's algorithm tolerable and balanced in calculation is a parallel structural design implementation. Recently, Dou et al in
20
proposed an innovative parallel design for accelerating Zuker's algorithm. The investigators in
20
utilized and built their techniques by dividing the Zuker algorithm matrix in the usual mode. Then, they submitted and distributed the subtasks as a multithreading procedure that can be calculated independently. This parallel scheme was implemented on the FPGA fine-grained hardware by proposing one master PE and multiple slave PEs with systolic array structure. In addition, the RNA researchers in
20
projected new methods for reducing the RNA energy lookup table size by 85%. These lookup tables should be loaded in the memory, as the RNA prediction algorithm requires to use them. Consequently, they implemented their parallel algorithm on 16 PEs on FPGA co-processors. The experimental results of this parallel scheme explained enhancement up to factor of 14× speed-up time comparing with the
RNA Parallel Taxonomy
Parallel taxonomy of RNA folding
In particular, the most popular RNA secondary structure prediction methods are the dynamic programming algorithms based on the MFE models. Herein, we proposed and described a general taxonomy to apply on the existing RNA parallel methods. This proposed taxonomy for the RNA parallel secondary structure methods is using four main phases. Three of them working as a different agents and the fourth one is the main RNA prediction algorithm, as shown in Figure 5:

Parallel taxonomy of RNA folding algorithms. (
The Input Checker Agent (INCA) receives the RNA primary sequence from the input device or reads it from a text file. Subsequently, the INCA checks the validity of this RNA by applying the valid nucleotides RNA bases in Watson-Wobble rules (
INCA transfers the valid RNA sequence to the parallel prediction RNA MFE algorithm (
Another parallel design is a combination of two existing pseudoknotted RNA secondary structures prediction methods.25,34 This design 17 used a parallel master-slave techniques based on the Beowulf cluster/hardware, as depictured in Figure 5 (c).
The last parallel scheme elaborated the 2D systolic array parallel by using FPGA in fine-grained hardware. This parallel paradigm was illustrated in Figure 5 (d). Actually, there were three various parallel RNA detection methods.18–20 These original RNA prediction methods were lately re-implemented on FPGA.
The Output Decision Maker Agent (OTDMA) checks the accuracy of the initial Output RNA Secondary Structure Production (ORSSP). It performs this checkable process by testing and comparing the first output “ORSSP” with the diverse known RNA structures (as a standard benchmark for testing and evaluating the RNA prediction method) in the RNA Databases (RNA-Db).53–57 If the OTDMA finds the quality of the new ORSSP is poor or low, it feeds the RNA sequence back to the INCA to re-start another round of detection with new and more intelligent constraints. Or else, the OTDMA finds the accuracy of the output RNA structure is high, it transfers the new ORSSP to the last agent, which called Final Result 2D Comparison Agent (FR2CA).
The FR2CA compares the final RNA secondary structure result with the known and existing structures in RNA-Db, to report the accuracy scale of predicted RNA. 58 In addition, the FR2CA agent measures the execution time and space complexities for the RNA prediction algorithm.
Evaluating existing RNA algorithms using comparative taxonomy
By using this proposed comparative taxonomy, as a classified comparison procedure for the existing parallel RNA prediction methods. The authors found that some of these RNA parallel prediction methods ran through all the comparative taxonomy phases. While, the other RNA prediction methods went through some steps of this proposed comparative taxonomy.
From the methodological point of view, the researchers explained and investigated a comparison procedure among these RNA parallel methods according to the proposed taxonomy in Table 2. The selected group of the previous RNA algorithms focuses on the parallel implementation of the existing RNA secondary structure methods.
The evaluating taxonomy for the parallel RNA secondary structure prediction approaches.

Discussion and Comparison
Scores of RNA researchers have introduced several evolutionary parallel blueprints for solving the RNA secondary structure problem. In this paper, the researchers discussed and compared these parallel RNA methods, as shown in Table 3. They have discussed in 22 an intensive RNA detection algorithms, as a first phase of the RNA secondary research in a Bioinformatics domain. The authors classified and compared the RNA approaches in two main groups and they presented the results in the two tables. In this study, the researchers extracted the well-known RNA sequential prediction methods in Table 1.
Comparison parallel algorithms for RNA secondary structure prediction.
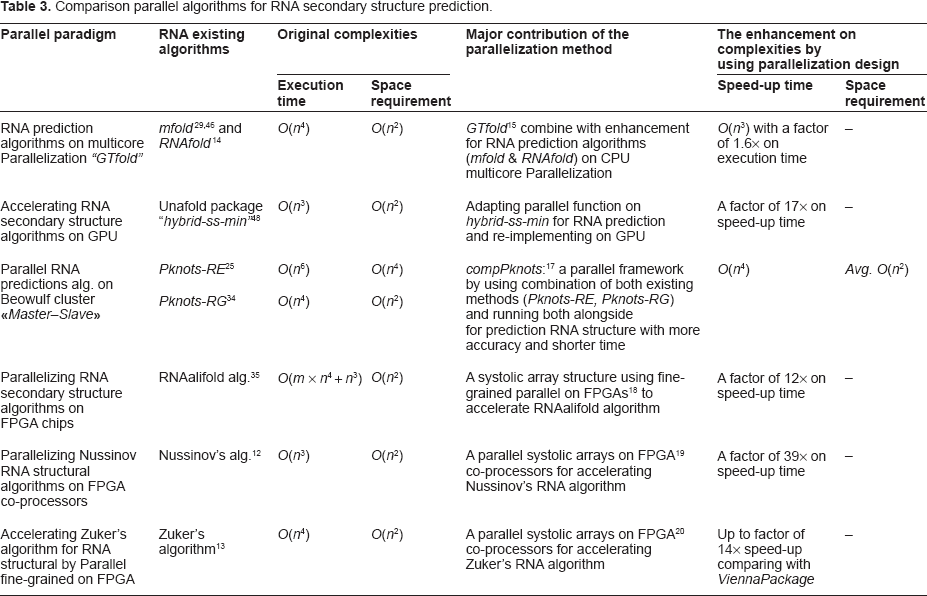
Then, in this paper, the authors focused their analysis on discussing and comparing the latest parallel RNA prediction efforts that have been pioneered in the RNA secondary structure folding domain. Consequently, from the comparison of the parallel RNA methods in Table 3, it could be noticed that only one research 17 presented a parallel design for the RNA pseudoknots type. While the others, proposed parallel designs for the RNA detection on stem-loops types. The comparison had also shown that the latest parallel algorithm 16 applied on the GPU, by utilizing the power of the NVIDIA card with the CUDA program. This method was implemented on the GPU to solve the RNA secondary structure problems. The high accessibility of the GPU card in the contemporary machines and the provided features for the developer to utilize GPU using high level languages like a C language environment; are the important motivations for the RNA research community to exploit the CUDA on the GPU.
Finally, the authors concluded that, implementing the RNA secondary structure prediction methods on parallel architectures has several significant benefits. The main advantage is reduced complexity, in both time and memory storage, comparing with the original RNA structure prediction algorithms. Also, the parallel RNA algorithms provided accurate results with better performance.
Conclusion
In this paper for the solving RNA secondary structure prediction problem, the authors presented the state-of-the-art of the RNA parallel methods. They introduced an intensive investigation on exhaustive up to date parallel RNA secondary structure prediction algorithms. Indeed, recently various methods and techniques for predicting RNA secondary structures have emerged. Most of these computational methods have faced with some complexities, from sequential implementation viewpoint. Therefore, re-implementing the existing RNA prediction methods by using parallelization architectures would result and contribute in solving the faced difficulties. The study concluded and showed that all of RNA parallel methods obtained better results, when compared to the sequential methods in terms of accuracy in one side and time/space complexities, in another side.
This research study was comprised in three trends: (
In addition, the researchers performed a scientific comparison among these enhanced RNA parallel methods with the previous existing methods. In the third part, the researchers proposed a new parallel taxonomy. Then, they applied the existing parallel methods using this taxonomy. Lastly, the researchers conducted a comparison procedure to evaluate these RNA parallel methods based on the proposed taxonomy, in terms of the taxonomy steps.
Consequently, this study of the RNA existing parallel methods proved that the parallelization performance of the algorithm is proportional to the method of the parallelization itself. Particularly, the comparison showed that the proposed RNA methods utilizing GPU capabilities result more promising outputs. Besides that, from the implementation point of view, the available open source Application Programming Interface (API) in a high level language in C environment could be considered as a positive point. These parallelizing RNA secondary structure prediction methods, showed a promising area for future RNA studies and for computational RNA bioinformatic researches.
Footnotes
Acknowledgment
This research was partly supported by the Universiti Sains Malaysia (USM) Fellowship awarded to the corresponding author. Also, it has been funded by USM Research Grant for the “A PARALLEL SWARM INTELLIGENCE BASED ALGORITHM FOR PROTEIN CONFORMATIONAL SEARCH” [304/PKOMP/638123]. The authors would like to gratefully acknowledge the help of Dr. Michael Liau Tet Loke (Editor, in the Postgraduate Academic Support Services (USM-P.A.S.S)) for proof-reading this paper.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.