
Abstract
The identification of nested motifs in genomic sequences is a complex computational problem. The detection of these patterns is important to allow the discovery of transposable element (TE) insertions, incomplete reverse transcripts, deletions, and/or mutations. In this study, a de novo strategy for detecting patterns that represent nested motifs was designed based on exhaustive searches for pairs of motifs and combinatorial pattern analysis. These patterns can be grouped into three categories, motifs within other motifs, motifs flanked by other motifs, and motifs of large size. The methodology used in this study, applied to genomic sequences from the plant species
Introduction
Repetitive sequences contribute to genome structure, function, and evolution. They are classified into transposable elements (TEs), tandem repeats, and high copy number genes.
1
TEs, defined as DNA fragments with structural and functional characteristics that allow movement throughout the genome, are the most abundant component of many genomes.
2
For example, almost 80% of the
The existing tools for the detection of TEs rely on the following four types of approaches: de novo, structure-based, homology-based, and comparative genomics (reviewed in the study by Bergman and Quesneville 6 ). In the de novo approach, the most common strategy detects all pairs of similar sequences at different locations within the analyzed input sequence, by comparing the input sequence to itself, concomitantly allowing the discovery and classification of repetitions such as TEs, tandem repeats, segmental duplication, and satellites. The use of this search method requires high-quality assemblies. 8 The second approach for TE identification uses prior knowledge of the common structures shared by different TEs. This method searches only for structures that have been previously reported. As reference databases do not include nested TEs, it is almost impossible to identify them using this approach. 9 The third approach involves searching for homology, usually based on a heuristic comparison within a sequence database. 10 In this case, if a TE is located inside another TE, the method reports only the internal element. Finally, the comparative genomics approach describes a group of methods that rely on neither homology nor structural features but detect new families of TEs based on the fact that TE transposition causes large insertions that can be detected with multiple sequence alignment. These methods search for insertion regions where multiple alignments of orthologous genome sequences are disrupted by a large insertion in one or more species. 11 Again, under this approach, a nested element will be reported only if it emerges from the comparison with some other genome. 6
Overall, these methods allow for relatively easy detection of TEs that lack mutations, insertions, or deletions that alter their sequences. However, in nature, individual TEs have great inherent biological complexity, with temporal divergence between sequences and the possible insertion of TEs inside other TEs, thus interrupting their structure. In this study, we propose a method to identify nested motifs (ie, a subsequence of the input sequence that is repeated two or more times and might correspond to TEs nested into other TEs) that may provide an advantage over conventional TE search methods that are not specifically designed to identify nested motifs.
The main objectives of this study were to identify perfect motifs repeated at any distance within the genome and detect the patterns of nested motifs. This approach is based on the fact that the LTRs located at each end of the TEs are almost perfect repeats.
Materials and Methods
Developing a tool to identify individual TEs is intricate given the inherent biological complexity of this problem, the divergence between the sequences, the presence of incomplete reverse transcripts, and the existence of nested sequences within other TEs. 12 Moreover, it is difficult to define the boundaries of an element since TE sequences with long deletions or insertions are also present in genomes. To address this complexity, we propose a de novo method that starts searching for perfect motifs at any distance. Then, an exhaustive search is performed between the locations where both motifs appear, to identify cases of a TE inside another one. In this context, this tool will allow analysis of the structures of insertions of different families of TEs by means of looking for specific features that can define the behavior of a TE family. Therefore, in addition to the initial identification of a list of putative motifs, the detection of TEs is addressed by a combinatorial pattern analysis approach.
Proposed approach
Our proposed approach is called Mamushka since its functioning resembles the Russian wooden nesting doll, a set of dolls of decreasing size placed one inside another. Mamushka uses Becher's et al algorithm 13 as the first preprocessing step. Becher's et al method performs a search for all perfect repeats in the genome based on the suffix array construction by Manber and Myers. 14 This algorithm uses the nucleotide sequence as input data, up to 500 million nucleotide bases, and a lower bound for the length of the patterns to be reported. The method returns a list where each element contains two lines; in the first line, the pattern is described together with its number of occurrences and its length, and in the second line the positions of each occurrence are listed. There is no upper bound for the length of perfect repeats and the occurrences can be at any distance from each other.
In the next step, a list (L) of
The final step of Mamushka is divided into two sub-algorithms called motifs within motifs (MWM) and motifs flanked by other motifs (MFM). For MWM, given two motifs, the objective is to determine whether one of the motifs is perfectly contained inside the other motif. P and Q stand for two motifs that belong to the L ordered list,
The second sub-algorithm searches for motifs flanked by other motifs. M, N, and target site duplication (TSD) denote three perfect distinct motifs taken from the L ordered list (Fig. 2B). The search determines whether N is flanked by other pairs of perfect motifs. M1 and M2 are defined to share motif M in different positions of the input sequence, as N1 and N2 share motif N and TSD1 and TSD2 share motif TSD. I and K denote the starting position of M1 and M2, and J and L denote their end; in addition, A and C denote the starting positions of N1 and N2, and B and D denote their final positions (Fig. 2B). Then, after verifying that M1I ≤ M1J ≤ N1A ≤ N1B ≤ N2C ≤ N2D ≤ M2K ≤ M2L, the algorithm continues the search for TSD elements. TSD1 should end at the General layout of the Mamushka method. The pipeline of the proposed new method to detect perfect repeat sequences is illustrated. The identified motifs are illustrated in the input sequence scheme. The frame encloses the steps followed by the method leading to a list of motifs within motifs and motifs flanked by other motifs. Mamushka rationale. (A) Motifs within other motifs. P and Q represent two motifs from the L ordered list, 

The use of big-O notation allows the classification of the algorithms based on how they respond, specifically describing the worst-case scenario. Both MWM and MFM are quadratic time algorithms O(
Additional technical details and software availability
Our Mamushka tool was written in Awk 15 and is available at the following website: http://lidecc.cs.uns.edu.ar/mamushka. To provide easy-to-use software, the code runs through a terminal console. The front-end of this script was created by invoking it from a website. Execution functions called EXEC were used to execute an external program, the Awk script. Then, for statistical purposes, an R-script 16 was programmed to show the distribution of the motifs and the number of the repetitions. After showing the distribution plot, the program allows the user to choose the range for the motifs, and motifs outside of this range will be discarded. After confirming the range, the program will perform another invocation of the code, this time calculating for motifs within other motifs, motifs flanked by other motifs, and large motifs within the stipulated size. This calculation will return three separate files that can be downloaded individually in LinDna format.
Validation of the Proposed Mamushka Method for the Detection of Nested Repetitive Elements
Given the deterministic and exhaustive exploratory features of the method, testing with artificial data would be trivial; accordingly, only real biological sequences were selected for the validation of Mamushka algorithm. By doing this, it was possible to demonstrate that the method can identify bona fide nested repetitive elements, even when the strategy searches for perfect repeated motifs. Thus, the experiment was conducted using DNA sequences from
Application example 1
The entire Frequency of various motif sizes. The frequency of the motifs was classified according to the size for (A) 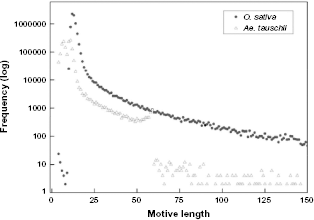
Thus, in rice chromosome 10, we found examples of motifs within motifs and motifs flanked by other motifs. In the first case, one of the repetitive sequences corresponds to the LTRs of the complete retrotransposon RLX_59520 from
In the second case, motifs flanked by other motifs, the inner sequence corresponds to a complete retrotransposon RLG_58802 from
Application example 2
In this example, 107 whole-genome shotgun sequences of
The distribution of perfect motifs showed a significant number of motifs with lengths ranging from 10 to 17 bp (Fig. 3). For
A sequence corresponding to a putative LTR retrotransposon flanked by another LTR retrotransposon in the whole-genome shotgun AOCO010454749 from
Matching regions are indicated by dotted lines, and the regions without a match are indicated by dashed lines in Figure 4.
Nested long terminal repeat (LTR) retrotransposons found using our Mamushka approach in whole-genome shotgun sequences. Mamushka allowed the identification of perfect motifs (white triangles) flanked by other perfect motifs (gray triangles). The BLAST analysis revealed that the identified sequences correspond to LTRs (light gray boxes). (A) The inner sequence showed identity to the LTR retrotransposon RLX_59520, flanked by the LTRs of the LTR retrotransposon RLG_60224. (B) The inner sequence showed identity to the LTR retrotransposon RLG_58802, flanked at both sides by two complete copies of the LTR retrotransposon RLG_5282. (C) The inner sequence showed identity to a putative LTR retrotransposon flanked by two halves of another retrotransposon, RLC66683.
These results were validated using RepeatMasker,6,19 which identified TEs by comparing them against known elements found in a database. The positions found by Mamushka constituted a subsequence of a sequence identified by the RepeatMasker.
Conclusion
We have developed a new method, termed Mamushka, for the de novo discovery of nested motifs. Our approach identifies repetitive patterns in DNA sequences that can constitute putative nested TEs, thus simplifying the process of detecting these elements. Mamushka recovers the nested patterns in three main steps. First, it performs a search for all perfect repeats in the genome using a suffix array construction. Next, all perfect repeats are ordered according to decreasing length. Then, two pattern recognition algorithms, MWM and MFM, are executed. For MWM, given two motifs, Mamushka determines whether one motif is perfectly contained inside the other motif. The MFM algorithm searches for motifs flanked by other motifs.
The Mamushka method is exclusively focused on the identification of perfect nested patterns. Nevertheless, this feature does not constitute a drawback for the applicability of our strategy with real biological data. Any mutated TE has a conserved subsequence up to the point where the mutation occurred, allowing Mamushka to identify it. Moreover, the search does not need to be conducted in both directions because Mamushka searches for a perfect repeat. These arguments are supported by the results obtained for two different plant species,
This pattern-matching approach can be combined with existing techniques to allow for the identification of nested repetitive motifs that cannot be found with existing methodologies. We intend to test the performance of Mamushka in other plant species in future work.
Author Contributions
Conceived and designed the experiments: JRR. Analyzed the data: JRR, IG. Wrote the first draft of the manuscript: JRR. Contributed to the writing of the manuscript: JRR, JAC, IG, VCE, IP. Agree with manuscript results and conclusions: JRR, JAC, IG, VCE, IP. Jointly developed the structure and arguments for the paper: JRR, JAC, IG, VCE, IP. Made critical revisions and approved final version: JRR, JAC, IG, VCE, IP. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
We also acknowledge Dr. Carlos Lorenzetti for his technical help in the development of the Mamushka tool website.