
Abstract
First, we identify InterPro sequence signatures representing evolutionary relatedness and, second, signatures identifying specific chemical machinery. Thus, we predict the chemical mechanisms of enzyme-catalyzed reactions from
Background
Protein signatures and InterPro
Enzyme function can be predicted using matches to sequence signatures based on models that classify proteins into families or predict the presence of characteristic domains or identifiable functionally relevant sites. Here, we describe a set of searches that we have conducted using signatures from InterPro,1–3 an integrated database, which combines predictive protein signatures from a number of different databases (Gene3D, 4 PANTHER, 5 Pfam, 6 PIRSF, 7 PRINTS, 8 ProDom, 9 PROSITE, 10 SMART, 11 SUPERFAMILY, 12 TIGRFAMs, 13 and HAMAP 14 ) into a single resource.
Each of the underlying databases contains sequence patterns that are biologically meaningful, for instance, corresponding to biochemical functions, homologous groups of proteins, or conserved domains. These are typically derived from models based on multiple sequence alignments, for example, hidden Markov models. The curators of InterPro identify those patterns, from the underlying source databases, that are considered sufficiently meaningful, informative, and reliable to be added as entries in InterPro. The process of integration involves curators identifying when signatures from different databases describe the same protein family, domain, or functional site. This is done by looking for multiple signatures that match the same set of proteins in the same region of the sequence. These signatures are then combined into a single InterPro entry. Grouping signatures into single entries such as this has the benefit of standardizing signatures to an extent, giving them consistent names and annotation, as well as removing redundancy. With each source database focusing on a particular niche in signature development, using all 11 databases together is extremely beneficial as it allows a diverse range of signatures to be combined.
An example is provided by the subclasses B1 and B3 of metallo-beta-lactamases, whose catalytic sites may well have evolved twice independently, but within the same evolutionary superfamily, to perform the same function by similar chemical mechanisms. 15 Many, though not all, metallo-beta-lactamases of both subclasses B1 and B3 hit PROSITE pattern PS00743, which includes catalytically important zinc-binding residues; many B1 lactamases also match PS00744, which includes another significant zinc-binding residue. In InterPro, these patterns are combined into signature IPR001018: beta-lactamase, class-B, and conserved site.
InterPro entries are of four kinds: families, domains, repeats, and sites. In the context of our work, we expect that most catalytic signatures in InterPro will be classified as
The 11 underlying databases each have different but complementary methods of calculating protein signatures. In general, the constituent databases describe matches to their identified patterns in terms of scores,
Enzyme Commission (EC) numbers and mechanisms
The long-established nomenclature for the classification of enzyme-catalyzed reactions is the EC number system. 17 EC numbers allow data to be computationally processed, but EC numbers classify neither the enzymes themselves nor their chemical mechanisms, focusing instead on the overall chemical transformation catalyzed by the enzyme. Therefore, if two different enzymes catalyze the same overall reaction, they will have the same EC number, whether or not they are structurally or evolutionarily related and regardless of the chemical mechanisms used. EC numbers classify enzyme reactions using a four-level system, with each succeeding digit giving a more detailed picture of the functionality of the enzyme. The first digit gives the class of the enzyme (eg, 4.-.-.- is a lyase). The second digit usually indicates the broad chemical nature of the reaction catalyzed (4.3.-.- is a lyase acting on a carbon-nitrogen bond). The third digit generally specifies the chemistry more precisely (here 4.3.1.- is an ammonia lyase), though the precise roles of the second and third digits vary by class. Finally, the full four-digit EC number indicates a particular enzyme-catalyzed reaction usually specifying the substrate (eg, 4.3.1.3 is a histidine ammonia lyase with L-histidine as its substrate).
Previous studies have found that using protein signatures to predict the EC numbers of enzymes is extremely effective.18–20 Cai et al.
18
found a subset accuracy in the range of 50.0%–95.7% for the prediction of enzyme families. De Ferrari et al.
20
achieved 87%–97% subset accuracy using InterPro signatures to reannotate several proteomes; this work was based on using a
Here, however, we are interested in identifying the signatures of catalytic machinery specific to a given chemical reaction mechanism, rather than an overall transformation. Hence, as in our previous work, 22 we predict enzyme mechanism rather than EC number. This also means, given the extensive effort required by experimentalists and annotators to confirm and record the exact mechanism of an enzyme, that we are limited by the size of the MACiE (Mechanism, Annotation, and Classification in Enzymes)23,24 database from which our enzyme mechanism assignments were taken; this database contains 335 entries of fully annotated enzyme mechanisms, each with at least one corresponding protein that is known to use this mechanism. Each entry contains detailed information on the individual steps, amino acids, and cofactors involved in each mechanism, all annotated from the relevant literature. The entries in MACiE differ from enzyme reactions as annotated by EC, because MACiE is able to differentiate between two reactions that share the same substrate and product but transform one into the other using a different chemical mechanism, whereas annotation by EC would indistinguishably describe such pairs of reactions with the same four-digit code. For instance, MACiE23,24 contains six separate β-lactamase mechanisms, all of which correctly correspond to the EC number 3.5.2.6. Nonetheless, the differences between these mechanisms, and especially between the serine-based and metallo-beta-lactamase mechanisms, are essential to understanding and countering antibiotic resistance.15,25,26
Homology and catalytic machinery
Matches to sequence signatures for enzymes contain two kinds of information. The first is that we can safely infer, from the shared sequence pattern or patterns, that the query sequence has common ancestry with enzymes whose functions are known or at least are sufficiently confidently asserted to be annotated in a database. The second is that the query protein sequence contains certain key residues positioned, in the sequence and presumably also spatially in the protein structure, to act as catalytic machinery. In most bioinformatics and function prediction contexts, these two types of information are mutually complementary and add weight to one another. Here, however, we want to separate them in order to understand the relative contribution to the overall predictivity that is made by each type of information.
Methods
Catalytic and non-catalytic signatures
Data were taken from MACiE 3.0, the protein data bank (PDB), 27 UniProtKB, 28 and InterPro v43.1 16 in September 2013. The raw dataset is made up of 540 proteins corresponding to 335 different MACiE mechanisms, 321 EC numbers, and 2,160 sequence signatures.
We want to distinguish between those (more numerous) sequence signatures whose matching corresponds to inference of homology and those (relatively few) representing a specific constellation of catalytic residues. While there is no perfect way of doing this, we identify catalytic and non-catalytic signatures by adopting MACiE's set of annotated sequence positions containing catalytic residues. These are defined as any residue that undergoes a change in electronic charge or covalent bonding or exerts an electrostatic or steric effect that facilitates the reaction. 29
We need to be able to identify the positions in our set of sequences that correspond to those annotated as catalytic by MACiE. Depending on the experimental method used to obtain the sequence, there can be slight differences between the type or number of amino acids found in what should be the same sequence. This usually appears at the start of sequences where one method has, for example, hydrolyzed off the initiator methionine, and so the final sequence is one amino acid shorter than in another version. For example, an entry in MACiE may state that the catalytic amino acids for mechanism X will be found at positions 3, 10, 25, and 67, but in the corresponding protein they are in fact found with an offset of +1 at positions 4, 11, 26, and 68. This offset is usually small, but in some cases, it was found to be as large as 90 residues. Allowing an automated process to search for a set of amino acids in offset positions is reasonable when there are three or more amino acids, and hence, the set is likely to be unique in the sequence, but when there are only one or two catalytic residues, this technique becomes somewhat unreliable. The issue with allowing variable offsets is ultimately a probabilistic one in the sense that as the allowed offsets become more generous, the probability of accidental matches increases. Therefore, we see it as, essentially, a trade-off between false positives (identifying a meaningless match because we used an offset too generously) and false negatives (missing a real match because we defined our offset criteria too tightly).
Having the gap between two residues as the only factor distinguishing these amino acids from hundreds of others in the sequence means that there is a possibility that the same amino acid combination may be found by chance (such a chance occurrence being unlikely to represent a viable instance of the catalytic machinery). To solve this problem, the offset was limited to 10 times the number of catalytic amino acids. This allowed the offset to be large when there were more catalytic residues but limited it to reduce errors when the number of catalytic residues was small.
In some cases, however, the amino acids were not found even when an offset was allowed. For example, in structure 1QDL 30 from the PDB 27 , the amino acids are expected to be in positions 57 (glycine), 84 (leucine), 85 (cysteine), 169 (histidine), and 171 (glutamic acid) in chain B. Leucine and cysteine are indeed found at positions 84 and 85, respectively, but the remaining three amino acids are not found in their expected positions. Glycine is found in position 56 with an offset of −1, while histidine and glutamic acid are found with an offset of +6 in positions 175 and 177, respectively. Examples such as these, which show conflicting offsets on manual inspection, were left out of the dataset. This was the case for only four proteins, so exceptions such as these did not have a significant impact on the dataset.
For the catalytic signatures, once the catalytic amino acids were located correctly, the next step was to create
The dataset was then refined for use in machine learning. Only the data corresponding to MACiE mechanisms that have two or more associated proteins were usable for machine learning. This is because a minimum of one protein is needed for training and another one for the test set; in the case of a
The numbers of catalytic and non-catalytic signatures, both in the raw data and in the refined set suitable for machine learning. This refined set had to contain at least two instances of each mechanism to permit training and testing, so all singleton mechanisms were removed in the refinement process. The total number of signatures is 556, which is unequal to the sum of 78 and 519 since some signatures are both catalytic and non-catalytic in different sequences.

While the proportion of signatures identified as catalytic was around 14% overall in both the raw and refined datasets, this proportion varied considerably depending on the source of the signatures. PROSITE signatures are of two kinds: profiles and patterns. A profile is one of the longer sequence features, usually identifying homology over a substantial section of sequence, whereas a pattern indicates the occurrence of particular conserved clusters of residues, considered to be functionally important, and typically 10–20 amino acids in length. Many catalytic site signatures are of this kind, and indeed, among the subset of our InterPro signatures that originated as PROSITE patterns, >50% appear as catalytic in our work. Among other sources of signatures, the proportion that is catalytic typically hovers around or below 10%.
As might be expected since they are typically sites rather than domains, families, or repeats, the catalytic signatures are generally much shorter. The average length for consistently catalytic signatures is 28 residues; for those signatures that are sometimes catalytic, it is 111 residues; and for consistently non-catalytic signatures, it is 226 residues.
Class labels
An instance in our datasets is composed of a protein identifier (a UniProt accession number), a set of attributes (matched InterPro signatures), and one or more class labels representing the MACiE mechanism(s) of the enzyme. A MACiE mechanism identifier corresponds to a detailed enzyme mechanism entry in the MACiE database modeled on one PDB structure and its associated literature. Figure 1 shows the sequences represented by InterPro signature sets, together with the associated MACiE mechanism labels. We also illustrate the closely related relevant information such as PDB codes, EC numbers, and domain names that can easily be associated with our data.
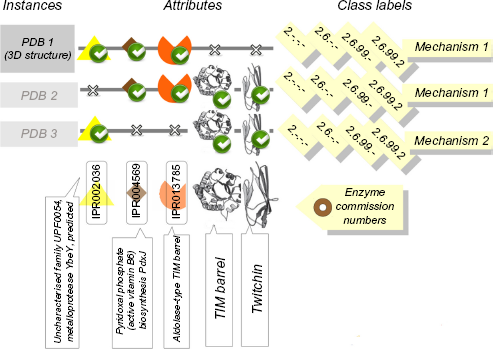
Illustration of the data, attributes, and labels used in this work. The sequences represented by InterPro signature sets, together with the associated MACiE mechanism labels, and also the illustration of the closely related relevant information such as PDB codes, EC numbers, and domain names that can easily be associated with our data.
Algorithm
Calculations were performed using the Mulan binary relevance
Multi-label learning methods can be split into two groups: problem transformation methods and algorithm adaptation methods. The first group of methods is algorithm independent and works by transforming the multi-label classification problem into multiple single-label classification tasks. The second group of methods alters the existing learning algorithms to allow them to handle multi-label data directly. BR-
The
Thus, each sequence is represented by the set of Inter-Pro signatures that are present within (ie, matched by) it. The distance between two sequences depends on the number of signatures that are present in one, and absent from the other sequence. Instances with exactly the same set of signatures will have the distance of 0. If the instances differ in one attribute, the distance will be 1; if the instances differ in
Sequences with zero signatures present could be problematic, as the algorithm described would see them as neighbors of the instance with the fewest attributes, though transferring the mechanism labels does not seem scientifically reasonable in such a case. To avoid this difficulty, two attribute-free and unlabelled dummy instances were added to the training data. Since MACiE annotated data are scarce, we use a leave-one-out cross-validation experimental design, where each prediction run is done using one enzyme as the test set and all other enzymes as the training set. Supplementary File 5 contains the Java source code to run the multi-label machine learning experiments and save the results.
Measures of classification success
We also use micro-averaged precision 

Results
MACiE mechanisms were predicted using the following: (1) only catalytic signatures, (2) only non-catalytic signatures, and (3) all available signatures, where catalytic signatures are those which disappeared under the
micro-averaged precision and sensitivity for catalytic and non-catalytic signatures.

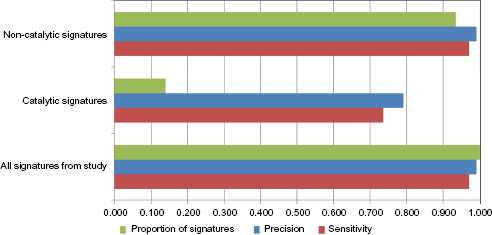
Classification performance of catalytic and non-catalytic signatures. The micro-averaged precision and sensitivity achieved by using the catalytic and non-catalytic sets and the proportions of our interPro signatures belonging to each group.
An analysis of all these results suggests that the prediction of enzyme mechanism is mostly by homology, as the sets of relatively long non-catalytic signatures containing homology information perform equally well as the full set, whereas the sets of short catalytic signatures perform markedly less well.
Thus, the homology clearly dominates the predictivity of our model, though it may well do so simply because evolutionary signatures are much more numerous and cover more of the dataset than catalytic ones and need not indicate that non-catalytic signatures are individually more powerful.
Discussion
We consider the short signatures to be likely to contain information about catalytic machinery, while long signatures contain information mostly concerning the evolutionary history of the sequence and also its possible homology with the query. We find that the 78 catalytic signatures taken alone do make some useful predictions. Nonetheless, the 519 non-catalytic signatures collectively do much better, their performance being identical to the values achieved by the full combined set of all signatures. Thus, adding the catalytic signatures would not improve the results obtained by the non-catalytic ones, and the non-catalytic signatures dominate the predictivity. The coverage of the catalytic signatures, that is sequences where catalytic signatures were present, in principle, could have been sufficient to correctly predict 170 mechanisms. Of these, 125 were correctly identified and 45 missed, while in addition 33 incorrect mechanisms were predicted. In contrast, the non-catalytic signatures correctly found 228 out of a possible 235 mechanisms and made only two incorrect assignments (Table 2).
Our previous paper on enzyme mechanism 22 contained a detailed analysis of false positive predictions, a pictorial representation of which was provided as supporting information with that work. The analysis of that study's false positives and false negatives showed that at least some of the false positive mispredictions involved closely related mechanisms or closely related protein families. For instance, our predictor confused anthranilate synthase (EC 4.1.3.27) and aminodeoxychorismate lyase (EC 4.1.3.38), which differ only at the fourth level of the EC classification. Similarly, it could not distinguish subclasses B1 and B3 metallo-beta-lactamases, which are usually considered distinct mechanisms, though they are similar and share EC number 3.5.2.6. In other cases, the similarities in EC number were less marked, but the mechanisms retained chemical features in common. We also looked at adding additional nonenzymes to the training data in that work, as expected the effect was to reduce the number of false positives at the cost of increasing the incidence of false negatives.
In the current work, both the full set and the non-catalytic set give a good balance between false positive and false negative predictions. The catalytic set, however, has substantially fewer signatures, and there is little surprise that in a significant number of cases it has insufficient information to make a correct identification, and hence records a false negative. What is less obvious is that there are nearly as many false positives, instances where the small sample of available signatures causes the predictor to misidentify associations. Looking at specific examples of false positives throughout the current study, a number of them involve confusing similar proteins or reactions and are fairly easy to understand and explain.
UniProt sequence P07598, actually associated with ferredoxin hydrogenase (MACiE M0127, EC 1.12.7.2), is misidentified as the adenylyl-sulfate reductase mechanism (M0123, EC 1.8.99.2). Both of these reactions are oxidoreductase processes involving iron–sulfur clusters, and the misprediction appears to stem from correctly identifying the binding sites for these clusters, but making an incorrect inference as to the reaction involved. The erroneous prediction in this instance comes exclusively from catalytic signatures.
Sequence Q13126 is actually associated with the S-methyl-5′-thioadenosine phosphorylase mechanism (M0244, EC 2.4.2.28), but our method misidentifies it as purine-nucleoside phosphorylase (M0017, EC 2.4.2.1). Given the high level of similarity between these two phosphorylase reaction mechanisms, and the structures which are both Rossmann folds, this is an understandable error resulting solely from non-catalytic signatures.
Another instance of our method by confusing two structurally similar proteins occurs with the sequence Q60099, which is actually
Another case of misassignment by confusing two Rossmann-fold enzymes occurs with sequence Q9ZGH3, which is actually a dTDP-glucose 4,6-dehydratase (M0228, EC 4.2.1.46), being assigned as an alcohol dehydrogenase (M0255, EC 1.1.1.1). The misidentification is made by catalytic signatures.
Although it is tempting to concentrate on the easily explicable errors in a case study approach, there are other mis-assignments that lack such clear and convenient rationalizations. The sequences O28603 and O28604, actually associated with the abovementioned M0123, are misclassified as proteasome endopeptidase complex (M0177, EC 3.4.25.1). There are some structural similarities between the adenylyl-sulfate reductase and proteasome endopeptidase complex, which are, respectively, 3- and 4-layer α-β sandwiches, though the reactions are not at all similar. The misidentification occurs through non-catalytic signatures alone.
Our method also misidentifies the same UniProt sequence O28603 of adenylyl-sulfate reductase (MACiE M0123, EC 1.8.99.2) as being an amine dehydrogenase (MACiE M0013, EC 1.4.99.3). Although there are superficial similarities between the reactions, which are both oxidoreductases utilizing nucleotide-like organic cofactors, there is no significant overall similarity between the proteins and this misprediction lacks a convenient explanation. We note that the sequence involved, Q28603 from UniProt, also failed to match its correct mechanism M0123 in our previous work. 22 All the signatures leading to this misprediction are non-catalytic and the false similarity is to the less catalytically important heavy domain of amine dehydrogenase.
In the current work, we look at the signatures indicative of homology and catalytic machinery in the sequence data only. In our previous research, the sequence information has proven successful in identifying both EC number 20 and mechanism, and in that case the addition of some three-dimensional information made little difference to the overall predictivity. 22 Nonetheless, it is interesting to consider how related studies operate using mainly or solely three-dimensional structural data. When present, homology can be readily detected from the three-dimensional structure, and indeed protein structure is widely believed to be more conserved than sequence for distant evolutionary relationships.36,37 However, such a study also has the capacity to detect the catalytic machinery through the location of three-dimensional templates38,39; this is a method that can identify mechanistic commonality even if the active sites are not related by homology, such as in the instance of the convergently evolved catalytic triads in subtilisin and chymotrypsin. 40 This kind of convergent evolution is the scenario in which it seems most likely that catalytic machinery information would be valuable for function prediction. Such catalytic information would only be available from three-dimensional features, since independent evolutionary inventions of, essentially, the same spatial arrangement of residues are not expected to recognizably leave similar sequence signatures.
It is also important to remember that the convergent evolution of catalytic function using, essentially, the same mechanism and machinery is the exception rather than the rule. Much more often, the convergent evolution to the same overall enzymatic function results in the development of a different chemical mechanism and the construction of quite distinct catalytic machinery. 41
Conclusions
These results show that our successful prediction of enzyme mechanism is mostly driven by homology rather than by identifying specific catalytic machinery. Indeed, limiting the information available to homology alone does not change the overall predictivity.
However, we need to be aware of the different numbers of catalytic and non-catalytic signatures. Thus, the longer profile-like features are much more numerous than the shorter catalytic site ones. In this situation, the sheer number and dataset coverage of the non-catalytic signatures allow them to contribute most to the model's predictive ability.
Author Contributions
Assisted with the curation of the dataset and analysis of the results and wrote the report that formed the basis of the first draft of this manuscript: KEB. Conceived the separation of catalytic and non-catalytic signatures, played the major role in gathering and curating the dataset, and executed the machine learning experiments: LDF. Conceived the original idea for this study and drafted the final version of the manuscript: JBOM. All authors reviewed and approved the final manuscript.
Supplementary Materials
Supplementary File 1
original_proteins_sequences.fasta
Supplementary File 2
mutated_proteins_sequences.fasta
Supplementary File 3
original_proteins_output.tab
Supplementary File 4
mutated_proteins_output.tab
Supplementary File 5
ml2db_code.tar.gz
These files give the sequences in FASTA format of the original (Supplementary File 1) and in silico-mutated (Supplementary File 2) protein sets, plus the outputs from InterProScan for the original (Supplementary File 3) and