
Abstract
Sickle cell disease (SCD) is a multisystem disorder characterized by chronic hemolytic anemia, vaso-occlusive crises, and marked variability in disease severity. Patients require transfusions to manage disease complications, with complements, directed by complement regulatory genes (CR1) and its polymorphisms, implicated in the development of alloantibodies. We hypothesize that CR1 polymorphisms affect complement regulation and function, leading to adverse outcome in SCD. To this end, we determined the genomic diversity of complement regulatory genes by examining single nucleotide polymorphisms associated with Knops blood group antigens. Genomic DNA samples from 130 SCD cases and 356 control Africans, 331 SCD cases and 497 control African Americans, and 254 Caucasians were obtained and analyzed, utilizing a PCR–-RFLP (polymerase chain reaction-restriction fragment length polymorphism) assay. Analyzing for ethnic diversity, we found significant differences in the genotypic and allelic frequencies of Sl1/Sl2 (rs17047661) and McCa/b (rs17047660) polymorphisms between Africans, African Americans, and Caucasians (P < 0.05). The homozygote mutant variants had significantly higher frequencies in Africans and African Americans but were insignificant in Caucasians (80.2% and 59.6% vs 5.9% for Sl1/2; and 36% and 24% vs 1.8% for McCa/b). With SCD, we did not detect any difference among cases and controls either in Africa or in the United States. However, we found significant difference in genotypic (P < 0.0001) and allelic frequencies (P < 0.0001) of Sl1/Sl2 (rs17047661) and McCa/b (rs17047660) polymorphisms between SCD groups from Africa and the United States. There was no difference in haplotype frequencies of these polymorphisms among or between groups. The higher frequency of CR1 homozygote mutant variants in Africa but not United States indicates a potential pathogenic role, possibly associated with complicated disease pathophysiology in the former and potentially protective in the latter. The difference between sickle cell groups suggests potential genetic drift or founder effect imposed on the disease in the United States, but not in Africa, and a possible confirmation of the ancestral susceptibility hypothesis. The lower haplotype frequencies among sickle cell and control populations in the United States may be due to the admixture and the dilution of African genetic ancestry in the African American population.
Keywords
Introduction
Sickle cell disease (SCD) is a complex multisystem disorder that presents with variability in disease pathophysiology and occurs in patients homozygous for the hemoglobin S (HbS) gene. It has a worldwide distribution1–4 with differing disease haplotypes, and is largely found in sub-Saharan Africa (Senegal, Benin, Bantu, and Cameroon haplotypes).5,6 The Arab-Indian haplotype is found in the Middle East, India, and parts of the Mediterranean.7,8 Among the African American population in the United States and populations in the Caribbean, SCD is commonly encountered with multiple and diverse disease haplotypes and clearly distinct and different from those in sub-Saharan Africa, 9 probably due to generations of admixture and genetic heterogeneity. 6 Oxygen is a major factor determining the degree of disease pathophysiology and the formation of the sickle-shaped red blood cells. These abnormally shaped cells obstruct blood flow, causing self-destruction of one's own red cells and the associated anemia. 10 Treating anemia and other disease complications require multiple transfusions, alongside the concomitant alloimmunization, which has been reported in some patient groups. 11 Biomarkers that are predictive of susceptibility to alloimmunization before the initiation of a transfusion regimen have been identified, 12 with complements potentially implicated in this process. It is imperative therefore to delineate the mechanism driving their activation and the pathway that directs their participation.
Complement receptor-1 (CR1) is a polymorphic glycoprotein, expressed predominantly on red cells, mainly functioning in the regulation of complement activity and the binding of C3b and C4b found on circulating immune complexes (CICs). Functional impairment of erythrocyte CR1 can interfere with the efficient processing of CICs, whose concentration in circulation is dependent on disease severity. 13 Due to its involvement in immune complex clearance, it has been associated with the severity of diseases such as malaria and systemic lupus erythematosus.13–16 CICs’ importance and their relationship to a variety of diseases, including sickle cell pathophysiology as well as intra- and interethnic delineation of disease haplotypes and phenotypes is therefore of great interest. Immune complex formation, under normal circumstances, is a process designed to facilitate normal functioning of the immune system, leading to the removal of CICs from peripheral circulation, which ultimately fails in some disease states, thereby initiating complement-mediated damage. Effective clearance of immune complexes, therefore, can be hampered by CR1 polymorphisms,17–20 which differ significantly between Caucasians and Africans and are shown to be associated with susceptibility to malaria infection or disease severity.21–24
Published reports have challenged the degree of protection conferred by Knops blood group polymorphisms in malaria infection.16,25,26 The co-evolution of malaria and SCD has been established, as demonstrated by reports showing that there are wide differences in Plasmodium falciparum infection rates and multiplicity of infection between carriers of the sickle cell trait (hemoglobin AS) and those with normal hemoglobin.27–31 Therefore, the possibility exists of increased selection pressure through single nucleotide polymorphisms (SNPs) that can exacerbate or ameliorate disease outcome.13,32 We believe that such protective polymorphisms in malaria could have similar evolutionary advantage in SCD, possibly assisting to delineate SCD pathophysiology. This study was designed to address the following questions: (1) What is the genetic diversity of African alleles of CR1 such as Knops blood group among and between SCD groups in Africa and United States? (2) Does this diversity follow an evolutionary pattern between these population and disease groups or any functional significance on disease severity? (3) If these diversities are significant, can we extrapolate the potential to interfere with the proper functioning of the complement system in SCD? To this end, we evaluated the association of these polymorphisms among SCD and healthy control populations and between Africans and Americans with SCD and analyzed interethnic differences in well-characterized and distinct ethnic control populations. We determined the genotypic, allelic, and haplotype frequencies of the CR1 SNPs (Kna/Knb, McCa/McCb, Sl1/2) in all groups.
Materials and Methods
Subjects
This study encompasses SCD patients (cases) and healthy control groups from the Centre de Recherche et de Lutte contre la Drepanocytose (CRLD), an SCD referral center in Mali. Ethical approval was obtained from the Institutional Review Board, Rochester Institute of Technology. Exemption from Federal definition of research with human subjects was granted, as the study uses de-identified, discarded samples. This research was conducted in accordance with the principles of the Declaration of Helsinki. Ethical approval of the original study in Mali, from which samples were obtained, was granted by the National Ethical Review Board, Mali. Patients gave their informed consent to participate in that study.
To be included in this study, patients had to meet criteria including diagnosis with SCD and hospital presentation during crisis or regular patient follow-up. Patient demographics and SCD analysis have been described earlier. 6 In brief, the SCD population from Mali is made up of 51.5% males and 48.4% females (mean age: 21 years; range: 1–51 years), predominantly of the Bambaran tribe. Healthy control populations from Africa and the United States are as described previously.6,33
Samples
Discarded blood samples collected into ethylenediaminetetraacetic acid (EDTA) tubes from 389 subjects from Mali (145 SCD patients and 244 healthy controls) were spotted onto Whatmann filter papers (GE Healthcare Sciences), and genomic DNA was extracted with the Qiagen Blood Mini Kit (Qiagen Inc., Valencia, CA), with some changes to the manufacturer's instruction. 6 Final elution volume was 100 μL, and the DNA samples were stored at −20 °C until further analysis. Genomic DNA samples from African American and Caucasian healthy controls (provided by Joann Moulds, Grifols, USA) were obtained from Shreveport, LA. African American SCD patients were recruited as part of the National Institute of Health-funded Cooperative Study of Sickle Cell Disease (CSSCD) (DNA samples provided by Betty Pace, Georgia Regents University, Augusta, GA).
Knops blood group genotyping
We selected the Knops blood group polymorphisms on the basis of their known association/interaction with malaria and disease severity in Africa and the protection conferred from malaria infection on individuals with hemoglobin S. A polymerase chain reaction (PCR) assay, which allowed the detection of all three Knops mutations, was utilized for genotyping.20,34 The primer sequences are as published (24 KnNde-5'ACCAGTGCCACACTGGACCAGATGGAGAACAGCTGTTTGAGCTT3’ and 25Rb-5'GGAGGAGTGTGGCAGCTTG3’), with the forward primer containing a deliberate mismatch for the detection of the Kna polymorphism. Two microliters of genomic DNA template was used for PCR amplification, with conditions altered to 50 μL final volume and amplified using the Lucigen EconoTaq Plus Green 2X Master Mix PCR system (Lucigen Corporation, Middleton, WI), as described. 32 The PCR cycling parameters followed published protocol, 34 and 5 μL of the amplified PCR product was evaluated on 2% ethidium bromide-stained agarose gels, visualized, and photographed. PCR product size (305 bp) was estimated with a TriDye 100 bp DNA ladder (New England Biolabs, Boston, MA), and size analysis was carried out with the Doc-It LS image analysis software (UVP Life Sciences, Upland, CA).
Restriction fragment length polymorphism (RFLP) assay
To detect the Knops blood group polymorphisms carried by CR1 [(Kna/Knb (rs41274768), McCa/McCb (rs17047660), and Sl1/Sl2 (rs17047661)], we made some slight modification to an earlier published protocol. 34 For Kna/Knb RFLP detection, 10 μL of PCR product was mixed with 1 μL of Nde I (20 U) and 5 μL of 1× CutSmart buffer (New England Biolabs) and incubated at 37 °C for two hours. For McCa/McCb, 1 μL of Bsm I (20 U) was added to 10 μL of PCR product and 5 μL of 1× CutSmart buffer and incubated at 65 °C for two hours. For Sl 1 /Sl 2 , 1 μL of Mfe I (20 U) was added to 10 μL of PCR product and 5 μL of 1× CutSmart buffer and incubated at 37 °C for two hours. The digestion products were analyzed on a 2% ethidium bromide-stained agarose gel, the band size was compared to a TriDye 100 bp DNA ladder (New England Biolabs), and size analysis was done as described above. 32 Samples heterozygous for each SNP were run on the gels as a positive restriction enzyme control. All PCR genotyping and restriction digestion were conducted anonymously, as described previously. 33 Briefly, restriction analyses were carried out by two investigators; 50% of amplified products were subjected to repeat digestion (third investigator) with 100% concordance.
Statistical analysis
A PERL script (http://www.perl.org) was written to facilitate analysis of genotypic and allelic frequencies of each variant and to convert the original data files to an EH program format. Exact tests for deviation from Hardy-Weinberg equilibrium (HWE) were performed, with SNPs to be rejected based on the recommended threshold of P < 0.001 in control individual. Differences in genotype and allele frequencies between populations were assessed using chi-square tests. Haplotype frequencies were estimated and tested for disease differences with the EH program (http://lab.rockefeller.edu/ott/programs).35,36 Differences in haplotype frequencies among the three populations were assessed with the EH program by likelihood ratio chi-square tests.
Results
We elucidated the genetic diversity of complement regulatory genes by examining the SNPs associated with Knops blood group antigens such as the Knops a and b (rs41274768), determined by a G (Kna) or A (Knb) (V1561M change). McCoy a and b (rs17047660) were determined by A (McCa) or G (McCb) (K1590E change) and Swain–Langley 1 and 2 (rs17047661), defined by an A (Sl1) or a G (Sl2) (R1601G change) among SCD and control groups in Africa (n = 130 and 356, respectively) and the United States (n = 331 and 497, respectively). In addition, we also determined interethnic diversification of these polymorphisms in healthy control population groups of Africans (n = 356), African Americans (n = 497), and Caucasians (n = 254). The demographics of the African SCD and control populations have been described previously, 6 as are the American population. 33 We utilized the PCR–RFLP assay, which has produced consistent results in the past.13,34 All polymorphisms were in HWE (P > 0.05).
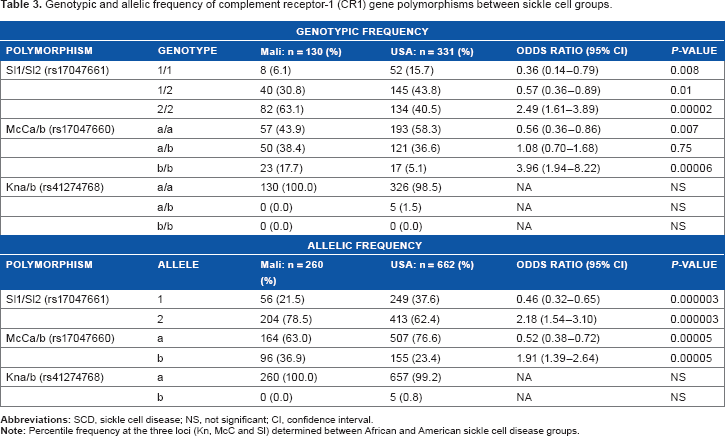
The most significant results from our study involved the Sl1/2 (rs17047661) and McCa/b (rs17047660) polymorphisms. Examining SCD cases and controls from Africa or the United states yielded only wild-type forms of the Kna/b polymorphisms, and hence were not included in our analysis. The distribution of Sl1/2 (rs17047661) and McCa/b (rs17047660) polymorphisms between SCD and controls showed no difference in genotypic (Table 1) or allelic (Table 2) frequencies among cases and controls, either in Africa or in the United States. However, we found a significant difference in both genotypic and allelic frequencies of the Sl1/2 (rs17047661) and McCa/b (rs17047660) polymorphisms between African and American sickle cell groups (Table 3). The homozygote mutant variant of the Sl1/2 (rs17047661) polymorphism occurred at significantly higher frequencies [63.1% and 40.5% (P = 0.00002) for African and American SCD, respectively (Fig. 1)]. The same observation was made for the McCa/b (rs17047660) polymorphisms, with genotypic frequencies of 17.7% and 5.1% [(P = 0.00006) in African and American SCD, respectively (Fig. 2)].

Genotypic frequency of Sl1/2 (rs17047661) polymorphisms between African and American sickle cell disease groups. Blue bars: African sickle cell disease; red bars: American sickle cell disease; wild-type homozygotes (Sl1/1); mutants (Sl2/2).

Genotypic frequency of McCa/b (rs17047660) polymorphisms between African and American sickle cell disease groups. Blue bars: African sickle cell disease; red bars: American sickle cell disease; wild-type homozygotes (McCa/a); mutants (McCb/b).
Genotypic frequency of complement receptor-1 (CR1) gene polymorphisms between sickle cell and control groups.

Allelic frequency of complement receptor-1 (CR1) gene polymorphisms between sickle cell and control groups.

Genotypic and allelic frequency of complement receptor-1 (CR1) gene polymorphisms between sickle cell groups.
We constructed haplotype groups using SNPs of the three loci [Kna/b (rs41274768), McCa/b (rs17047660), and Sl1/2 (rs17047661)] under investigation. We found eight haplotype groups that were formed but could find no difference in haplotype frequencies among SCD and control groups, either in Africa or in the United States (Table 4). Among the Africans, the most common haplotype combines the wild-type homozygotes (haplotype 1: Sl1/1, Kna/a, and McCa/a) for all three polymorphisms (42% and 44% for SCD and healthy controls, respectively), followed by haplotype 3 (36% for both SCD and control groups). Haplotypes containing the homozygote mutant form for all three polymorphisms or a combination of such (haplotypes 2, 4, 6, and 8) were not found. Similar observation were made among Americans, with haplotype 1 found with the highest frequency for all three polymorphisms (39% and 36% for SCD and healthy controls, respectively), followed by haplotypes 5 (37% and 39%, respectively) and 3 (23% for both case and control groups). Haplotypes containing the homozygote mutant form for all three polymorphisms or a combination (haplotype 2, 4, 6, and 8) were not found as well.
Estimated haplotype frequencies of the three SNPs between African and American sickle cell and control groups.

To clarify the possibility of ethnic diversification in genotypic and allelic frequencies, we examined the distribution of the Knops blood group polymorphisms in control populations from three ethnically classified groups (Africans, African Americans, and Caucasians). We found a significant difference in the genotypic (Figs. 3 and 4) and allelic (Table 5) frequencies of Sl1/2 (rs17047661) and McCa/b (rs17047660) polymorphisms between the ethnic groups. The homozygote mutant variants had significantly higher frequencies among Africans, followed by African Americans, but they were insignificant in Caucasians (64.9% and 37.2% vs 0.8% for Sl1/2; rs17047661 and 13.5% and 5.8% vs 0% for McCa/b; rs17047660). Haplotype groups, similar to that constructed for case and controls, were analyzed, and significant interethnic difference in the haplotype frequencies between Africans, African Americans, and Caucasians was found (Table 6). The most commonly encountered haplotype (haplotype 1) combined the wild-type homozygotes for all three polymorphisms (19%, 40%, and 94% for Africans, African Americans and Caucasians, respectively), followed by haplotype 5 (44%, 37%, and >1% for Africans and African Americans, respectively) and haplotype 7 (36% and 23% for Africans and African Americans, respectively). Haplotypes 5 and 7 were absent among Caucasians, while haplotypes 2, 3, 4, 6, and 8 were not found among the three ethnic groups.

Genotypic frequency of Sl1/2 (rs17047661) polymorphisms between ethnic groups (control populations). Black bars: Africans; blue bars: African Americans; red bars: Caucasians; wild-type homozygotes (Sl1/1); mutants (Sl2/2).

Genotypic frequency of McCa/b (rs17047660) polymorphisms between ethnic groups (control populations). Black bars: Africans; blue bars: African Americans; red bars: Caucasians; wild-type homozygotes (McCa/a); mutants (McCb/b).
Allelic frequency of complement receptor-1 (CR1) polymorphisms in ethnic groups.

Estimated haplotype frequencies of SNPs in the CR1 gene in diverse ethnic groups.

Discussion
The genetic diversity of Knops blood group antigens determined by examining SNPs associated with these genes among SCD and control groups from Africa and the United States is reported. In addition, the potential role for interethnic variation in the distribution of these polymorphisms and implications for case management and disease pathophysiology are discussed. Our choice of these three sites is based on multiple reports (including ours) that have shown association/interaction of Knops blood group polymorphisms with malaria in Africa.13–16,20 SCD is highly prevalent in malaria endemic areas because of selection pressure that favors individuals with the recessive hemoglobin S, a major contributory factor to malaria resistance in this population.27–31 It is now accepted that SCD is a genetic mutation that arose as a protective mechanism against the scourge of malaria in West Africa. Considering this, and the large amount of CICs associated with both disease states 20 (SCD and malaria infection), it is imperative and scientifically necessary to examine the role of CR1 polymorphisms in SCD. The emergence of human genome sequencing and large-scale population-based studies have led to an increase in disease association studies utilizing gene polymorphisms that are thought to direct disease outcomes or clinical endpoints.
Among the cases and controls, there was no significant difference in either genotypic or allelic frequencies of Sl1/2 and McCa/b polymorphisms among SCD and control groups either in Africa or in the United States; nor was there any difference in haplotype frequencies among groups. This is rather surprising, since SCD is expected to have a selective pressure on the CR1 polymorphisms in the African population but not in the American population. This observation implies that these genes could potentially be stable and conserved within a population but not between populations. That is, an intrapopulation conservation and an inter-population diversification cannot be clarified by observations among SCD and control groups but by the significant diversity in the frequencies of the McC and Sl homozygous mutant variations between SCD populations in Africa and the United States. This difference reveals the potential changes imposed on the disease in the United States but not in Africa. This could possibly aid in our understanding of how inherited defects attributable to genetic drift/founder effects can direct disease susceptibility or outcomes in diverse populations, with respect to infectious and chronic disease, and in subpopulations of cases within a broad range.37–55 This is a confirmation of the ancestral susceptibility hypothesis, which states that disease susceptibility alleles are ancestral while derived variants are protective. That is, SCD in the United States (a new and nonnatural environmental niche) has become adapted to conditions of the New World, thereby imposing protective founder effects on sufferers not seen in ancestral groups. 56 The lack of haplotype difference among groups either in Africa or in the United States is noted, but the lower haplotype frequencies found among SCD and control populations in the United States compared to their African counterparts reveal the degree of genetic admixture in the American population36,57,58 and resultant dilution of African ancestry in the African American population.
Global distribution of SCD overlaps regions where P. falciparum infections are endemic, and hence the assertion that SCD mutation arose as a protective event against malaria infection.59,60 Published reports on the association between CR1 polymorphisms and malaria infection in Africa abound, although these studies have come up with conflicting conclusions.13,19,23,25,26,61 A recently published report 16 states there is no association between human genetic polymorphisms in the Knops blood groups and a protective advantage against P. falciparum malaria in Ghana, and that the theory of selective pressure on CR1 is probably unrelated to malaria but to other infections. 62 Our report confirms this theory and extends our hypothesis of CR1 gene-selective sweep in new climes.36,63–65
The difference in Sl1/2 and McCa/b polymorphisms between African, African American, and Caucasian groups as well as the extensive interethnic haplotype frequency is a confirmation of previous reports,20,21,23 summarized in Hansson et al. 16 The disparity in haplotype frequencies between the African and African American population vis-a-vis an Afro-Brazilian population 66 reveals that phenotypic similarity is not sufficient to decipher potential genetic diversities that might have implications for disease outcome. The higher frequency of CR1 mutant variants in Africa but not in the United States reveals a potential pathogenic role, possibly associated with complicated disease pathogenesis in the former and potentially protective in the latter. The possibility that these polymorphic variants could be contributors to alloimmunization12,67–70 or post-transfusion hyperhemolysis 71 in American but not African SCD deserves further scrutiny. The mutant variant is almost nonexistent among Caucasians for both McC and Sl polymorphisms, confirming previous reports and conferring advantage seen elsewhere but not in Africa.7,72
The significant difference in estimated haplotype frequencies between the three populations confirms our hypothesis that the observed interethnic disparities have a potential effect on disease outcome and clinical endpoints vis-a-vis SCD, and that the more complicated and severe cases are expected among Africans while milder forms of the disease would be found in the United States. Teasing out these polymorphisms in larger African and American SCD groups, as well as individual or associative clinical endpoints, would be a perfect way to decipher the significance of complement regulatory genes in sickle cell variability and disease pathophysiology.
Author Contributions
Conceived and designed the experiment, and optimized protocols: BNT. Carried out the clinical sample collection and sickle cell genotyping in Mali: AG, DAD. Carried out DNA extraction, genotyping, and restriction digestion: BNT, KCD, JAN. Drafted the manuscript, designed the tables and figures: BNT, IGI. Carried out the statistical analyses and revised the manuscript: BNT, IGI, YL. All authors read and approved the final version of the manuscript.
Footnotes
Acknowledgments
We are very grateful to Joann Moulds, Grifols, USA, for supplying the African American and Caucasian control DNA samples. Special gratitude is due to Betty Pace, Georgia Regents University, who graciously provided the African American sickle cell DNA samples, collected as part of the Cooperative Study of Sickle Cell Disease. Travel awards through the Provost Fund (KCD) and the Federation of American Societies for Experimental Biology Maximizing Access to Research Careers (JAN, BNT) are acknowledged. This work was presented at the Annual Biomedical Research Conference for Minority Students (ABRCMS), San Antonio, TX, USA, during November 12–15, 2014.