
Abstract
Major facilitators of water movement through plant cell membranes include aquaporin proteins. Wheat is among the largest and most important cereal crops worldwide; however, unlike other model plants such as rice, maize and
Introduction
Bread wheat, an allohexaploid plant also known as
On the basis of sequence similarities, plant aquaporins have been classified into 4 subfamilies: the plasma membrane intrinsic proteins (PIPs); tonoplast intrinsic proteins (TIPs); nodulin26-like membrane intrinsic protein (NIPs); small membrane intrinsic proteins (SIPs) and basic membrane intrinsic proteins (BIPs).11,12 There are a number of studies focused on the
Aquaporins exhibit a typically conserved structure with 6 transmembrane helices (TMHs; H1-H6), connected by 5 loops (loops A–E), and two Asn-Pro-Ala amino acid motif. This conserved NPA motif is found to confer substrate selectivity for molecularw transport. 13 The expression profile of each aquaporin gene family is regulated differentially. Javot and Maurel's 20 studies revealed that AQPs expression level was highly abundant in roots during soil water uptake.
Promising observations from the evaluation ofw aquaporins among stress resistance or sensitive plants, such as drought susceptible and drought tolerant wheat cultivars,
22
other crop cultivars,
23
or stressed EST libraries,
24
clearly indicate that aquaporins would be important for water uptake, transport and identification or development of any stress tolerant genotypes of crop species. The present work tackles challenges for development of functional marker (FMs) from sequence polymorphisms present in allelic variants of an aquaporin gene. FMs precisely distinguish alleles of a targeted gene, and are modern molecular markers for marker-assisted selection in wheat breeding.
25
Improvement of productivity of wheat cultivars under drought conditions has become one of the important breeding program objectives in wheat. The performance of genotypes under drought conditions is largely attributed to genetic variations, mostly at the single nucleotide level. Therefore, an
An enormous deal is known about aquaporin proteins in several plant species; however, little information is available about the aquaporin gene family in wheat. This may be due to the unavailability of its complete genome sequence, as well as the allohexaploid nature of the genome, which is proving to be for gene identification and analysis. Previously, 35 PIP and TIP aquaporin genes have been identified in wheat as reported by Forrest and Bhave,
11
as well as Yousif and Bhave.
12
Determining and understanding of the molecular mechanisms underlying the response to abiotic stress responses is required for genetic improvement of wheat for stress tolerance.
23
Although
Materials and Methods
Database sources
The
Bioinformatics analysis
Open reading frames (ORFs) were generated by ORF finder(http://www.ncbi.nlm.nih.gov/gorf). Sub-cellular localization prediction of each predicted TaAQP was carried out using WoLF PSORT (http://wolfpsort.seqcbrc.jp) and TargetP 1.1 (http://www.cbs.dtu.dk/services/TargetP). 2D and 3D structure alignment was performed using MATRAS (http://strcomp.protein.osaka-u.ac.jp/matras/). Conserved domains and signature pattern analysis was performed using SMART (http://smart.embl-heidelberg.de/) and Scanprosite (http://prosite.expasy.org/scanprosite/) programs.
Phylogenetic tree construction
Amino acid sequences of 67 gramineous PIPs, TIPs, SIPs and TIPs, including 20 from rice (
Analysis of expression profiles
The expression profile was determined by analyzing the EST counts based on UniGene of

In silico mining of SNP
Aquaporin ESTs were downloaded from Unigene and were cleaned to remove contaminating sequences. Vector sequences and other contaminations were identified by using the VecScreen web server (http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html). Poly-A/T tails were completely trimmed by the EST trimmer Perl program. After pre-cleaning, EST sequences shorter than 50 bases were discarded. Furthermore, low complexity regions were masked by using Repeat Masker (http://www.repeatmasker.org/). ESTs clustering and SNP identification as performed on the Seqman module of DNAstar software (DNASTAR, USA). A Perl script was written to parse Indel data from output results.
Validation of SNPs
The allele-specific primers were designed from the position of base transition. The rationale in designing the primers was based on the premise that the 3′-terminal positions ought to be unique among the known wheat genomic sequences. The primers were designed from consensus sequence of contigs which have the candidates SNPs by Primer3 (http://frodo.wi.mit.edu/cgi bin/primer3/primer3_www.cgi) with Tm 55 °C–65 °C, and PCR product size 175 bp. Polymerase chain reaction (PCR) was performed in a 25 μL volume containing 100 ng of genomic DNA, 2.5 μL of 10 xPCR buffer, 200 μM of each dNTP, 0.2 μM of each primer and 1.0 unit of
3-D model generation and validation
The model of
Results and Discussion
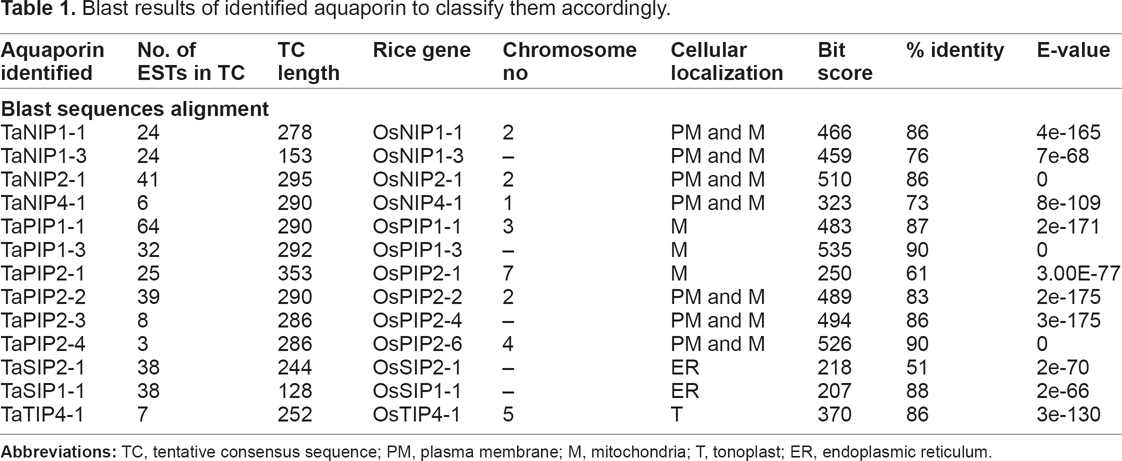
Identification and characterization of aquaporin genes in wheat
ESTs have proven to be an excellent source for gene discovery, molecular marker discovery and gene expression analysis. To ascertain aquaporin genes in the wheat genome, wheat's UniGene database, Ta.seq. all, which contains 56,943 entries, was obtained. Aquaporin families from
Blast results of identified aquaporin to classify them accordingly.
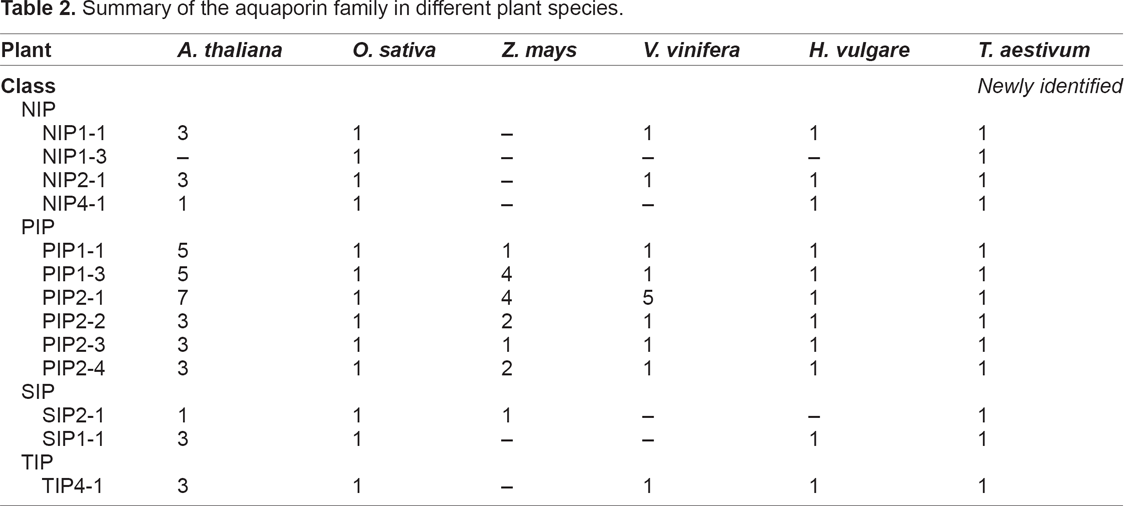
Comparisons of aquaporin genes in selected plant species
For comparative analysis, we used aquaporin genes of rice, barley, wheat,
Summary of the aquaporin family in different plant species.
Sequence alignment and conserved motifs in wheat and rice
The sequence and structural alignment of deduced amino acid sequences of wheat and some well-described AQPs from selected plant species showed a high degree of homology (Fig. 1). Considering the high homology between aquaporin subgroups irrespective of species, these groups may have evolved before the divergence of higher plants. It was observed that all the aquaoprin subfamilies have a tendency to have alpha (a) helical structure followed by a beta (β) sheet. PIP2 genes are highly homologous as compared to PIP1 and other aquaporin subgroups (Fig. 1B and C). Functional studies of AQPs showed that PIP2s reveal high osmotic water permeability in contrast with PIP1 members that show lower or no water permeability when expressed in

Multiple sequence alignment using amino acid sequence of (A) TaNIP1-1, TaNIP1-3, TaNIP2-1, TaNIP4-1 (B) TaPIP1-1 and TaPIP1-3 (C) TaPIP2-2, TaPIP2-3 and TaPIP2-4 (D) TaTIP1-4, TaSIP1-1 and TaSIP1-2 from different cereal crops. Conserved amino acids are shown in black boxes. Protein structural features are indicated above the alignment. Alpha helix and beta strands are elements of secondary structure represented as rods and arrows.
Computational prediction revealed up to 6 transmembrane helices with 21 amino acid residues in each helix among all 13 deduced amino acid sequences of TaAQPs as shown in Supplementary Figure 1. Predicted amino acid sequences of the aquaporin gene family revealed an NPA motif (Asn-Pro-Ala) that was extremely conserved.11,12 Two NPA motifs were predicted in PIPs, NIPs, TIPs, and one in SIPs (Supplementary Fig. 2). The NPA boxes and adjacent residues are considered essential for water transport activity.
31
The first NPA box is located in the first cytosolic loop and the second NPA box is located in the third extracellular (or vacuolar for TIPs) loop. Substrate selectivity for molecular transport is recognized by the presence of an NPA box.
32
The helical region is of particular importance for AQP structure because they contain the conserved NPA motifs that are functionally important for water channels. The C-terminal region of TaPIPs and NIPs encompasses conserved phosphorylation sites KXSXXR/K (Supplementary Fig. 2). Phosphorylation of the C-terminal serine may regulate aquaporin activity in response to an osmotic signal.
18
An increase in aquaporin activity following phosphorylation was demonstrated in α-TIP and PM28A in oocytes.
33
AQPs belonging to PIP1 subfamily encode polypeptides of 244–292 amino acids in length with 92.47% (TaPIP1-1 and TaPIP1-3) shared sequence identity in wheat. The length of the PIP2 (PIP2-3) ORF ranged from 170–320 amino acids, which had a relatively high sequence similarity of 88% with PIP2-2. In addition, PIP2-2 and PIP2-4 shared 80% amino acid sequence identity with each other. In general, lengths of estimated ORFs were quite different between PIP1 and PIP2. The predicted single polypeptide of TIP1 was 252 amino acids in length. However, predicted SIP1-1 polypeptide ORFs ranged from 128 to 244 amino acids that shared 55% sequence identity (see Supplementary Table 1). NIPs were divided phylogenetically into 4 different subgroups (NIP1-2, NIP1-3, NIP2-1 and NIP4-1) with ORF lengths ranging from 245 to 250 amino acids. However, TaNIP2-1 and TaNIP4-1 (26%) showed minimum identity. After detailed fingerprinting analysis of different classes of putative aquaporin, we found that these AQP proteins have a signature motif identical to phosphokinase C, tyrosine kinase and casein kinase II proteins, as well as N-myristoylation site and MIP family signature sequences SGxHxNPA shown in Table 3. Specific signature pattern scans were identified to be PKC_PHOSHO_SITE, N-Myristoylation site, MIP signature and CK2_PHOSPHO_SITE families of 3, 6, 4 and 9 amino acids in length and present in all 4 classes of aquaporin. These families have a more or less significant relationship with regulation and metabolism. Plant protein kinase C (PKC) cascades are likely to be involved in mitogen-activated signaling pathways, cellular regulation and metabolism, gene expression, metabolism, motility, membrane transport, and apoptosis.
34
Previous studies have reported that Casein kinase II, a selective protein kinase, is possibly involved in circadian clock regulation, photoperiod sensitivity
35
and various regulatory processes of plants. In eukaryotes, N-myristoylation (N-MYR) plays an important role in cell physiology, alters the lipophilicity
36
of the target protein and facilitates its interaction with membranes, thereby affecting its subcellular localization. N-MYR in plant cells has a critical role in controlling membrane-signaling pathways that lead to specific plant immunity.
37
For instance, in
Fingerprint analysis result for different aquaporin classes.


Phylogenetic tree was constructed using the neighbor-joining method and diagrams drawn with MEGA4. NJ method used the multiple sequence alignment generated by ClustalW to generate the tree. Bootstrap values are indicated against each branch. Phylogenetic tree showing the 4 clusters of PIPs, TIPs, NIPs and SIPs. The 13 wheat AQPs are compared with all the PIPs as well as the TIPs, NIPs and SIPs from
Sub-cellular location of the predicted AQPs
AQPs were characterized in this study and a link with AQPs subcellular localization was established. Identified TaPIPs, TaNIPs, TaTIPs and TaSIPs were figured out, to contain a signal peptide localized in various places; ie, mitochondria (TaPIP1-1, TaPIP1-3 and TaPIP2-1), tonoplast (TaTIP4-1), plasma membrane (PM) and mitochondria (TaNIP1-1, TaNIP1-3, TaNIP2-1, TaNIP4-1, TaPIP2-2, TaPIP2-3 and TaPIP2-4), and endoplasmic reticulum (TaSIP1-1, and TaSIP1-1; Table 1). Proper sub-cellular localization represents a mechanistic regulation of aquaporin activity that possibly relies on its ability to form multimers between members of different subgroups.
39
In consistent with previous reports, TaPIPs proteins were found in PM and mitochondrial sub-cellular compartments, which are identical to those predicted in ice plants (
Phylogenetic relationships of aquaporin family genes in different plant species
To understand the evolution and conservation of crop species, elucidation of the evolutionary relationships is a crucial step.
41
Earlier, Zardoya et al
42
established a phylogenetic framework for the aquaporin family in eukaryotes. In order to systematically classify the putative TaAQP, a phylogenetic tree was constructed using bootstrap analysis based on multiple sequence alignments of the proteins sequences of
Expression profile analysis of aquaporin genes
It has been suggested that the aquaporin gene family expression pattern in response to abiotic stress in different plant tissues at various stages has been detected in various plants. To demonstrate the utility of digital expression analysis of numerous genes across various tissues, we performed gene expression analysis of the wEST collection.
43
The expression profiles varied with aquaporins subfamilies. 5 genes from PIP (PIP1-1, PIP1, PIP2, PIP2-3, and PIP2-7), 6 genes from TIP (TIP2-2, TIP2-1, TIP2-3, TIP1-2, TIP3-1 and TIP4-3), 3 each from NIP (NIP1-1, NIP1-3, and NIP3-2) and AQPs (AQP5, AQP3, AQP4, AQP2, AQP7) were analyzed to compare the abundance of mRNA transcripts in various tissues. The aquaporin family genes from

Distribution of

Tissue specific expression of the aquaporin genes profile was determined by analyzing the EST counts based on UniGene.
In silico identification and validation of SNP
The availability of huge plant sequence data in the public domain is a rich resource for discovery of high-quality SNPs using bioinformatics pipeline. EST databases are an abundant source of SNP markers. 45 Recently, Mondini et al 46 identified SNPs involved in drought and salt stress tolerance in durum wheat. In this study, 1381 aquaporin ESTs were assembled into 32 contigs. Assembly of these cleaned ESTs was done with stringent parameters (match size = 40, sequence length = 100, maximum expected coverage = 40 and match percentage = 95). Only contigs containing a minimum of 4 and maximum of 80 members with 4 different cultivars were analyzed further. Not enough information can be gained from contigs < 4 EST and it becomes difficult to view and edit contigs > 80 ESTs. SNPs were declared only when there was no mismatch, no gaps before and after putative SNP site; in addition, the alternative base in the consensus sequence was present at least more than twice in an alignment. 6 SNPs from 9 contigs were taken into consideration with SNP score ≥ 40 and SNPs in the start and end of the alignment were ignored (Table 4). In current investigation, we found only a few SNPs because less EST sequences were available for the aquaporin gene. The allele-specific marker (Supplementary Table 2) was used for amplification of DNA fragment in 38 wheat genotypes (Supplementary Table 3), representing a core set. 47 20 genotypes resulted in the presence of a band of ~170 bp fragments (Fig. 5), and the remaining 18 genotypes did not yield any amplification. 1 SNP was found between these genotypes. The primer is amplified mostly in drought tolerant genotypes, based on the reported phenotypic data analysis (BS Tyagi, personal communication). This is the first report of a PIP-derived SNP marker in wheat that can be used by the breeders for improving tolerance to drought and high temperatures in wheat breeding programs.
Details of ESTs and predicted SNPs in aquaporin genes.


PCR analysis using the SNP marker. List of genotype was mentioned in Supplementary Table 2. The size of the amplification product is shown on the left. Absence of a band indicates the specific sequence is absent in wheat genotype. Lane 39 is a negative control, which used H2O as the template.
3D model prediction and validation
After establishing functional annotation, we have now extended research on the structural background of aquaporin protein in order to gain more insights. Lack of theoretical 3D structure of TaAQP protein motivated us to carry out this study. Therefore, we modeled wheat PIP (Accession no: AAM00368) using BLASTP identified the crystal structure with PDB ID: 2B5F as templates. Query sequences showed 74% sequence identity with the template with an E-value of 1e-59 (Fig. 6A). Only 1 (0.4%) out of 292 residues was present in the disallowed region whereas another 4 (1.7%) residues were present in the generously allowed regions of the Ramachandran plot, respectively (Supplementary Fig. 3). Similarly, despite of having ~74% sequence homology amongst template and modeled proteins, the tertiary structure was also found to be comparable as indicated by a root mean square deviation of 0.60 Å. Holistically, the modeled tertiary structure of

(A) Pairwise alignment of aquaporin family gene from
We believe that the present findings illustrate more insights into the structure-function role of AQPs protein in molecular terms. In order to understand the function of individual MIPs in maintaining water homeostasis, it is necessary to carry out knock-out experiments and promoter analyses, as well as substrate specificities under various physiological conditions in relation to water balance and nutrient uptake in wheat and other plant systems. Current biotechnology and bioinformatics tools may identify and characterize genes in their respective subclasses. As a model plant, and having a great synteny with the grass family with respect to gene structure, the information generated about the aquaoporin gene family in wheat will also provide a platform for predicting the function of genes of crops whose genome sequences are in their infancy.
Conclusions
The significance of the multigene family of aquaporin transmembrane proteins is emerging from studies aimed at optimizing water and nutrient use efficiency. Complete set of AQPs have been identified and classified in some agriculturally important crops with well-determined genomic sequences. Though the global importance of wheat as a most important cereal grain in world trade is well established, its AQPs remain less studied to date because of the lack of wheat genomic sequence information. The goal of this study was to identify new members of the aquaporin family in the wheat genome. Our combined approach enabled us to identify a total of 13 new aquaporin genes in wheat that showed significant sequence identity (>50% identity) with those from rice. A number of motifs and signature patterns were identified that are related to subcellular localization and functional annotation. Characterization of SNPs in candidate genes for drought tolerance such as aquaporin is a promising approach for identifying alleles that are associated with drought phenotypes. The aquaporin gene obtained in this work thus provides further tools for the physical and genetic mapping of these important genes, for identifying their chromosomal locations or genetic linkage to water homeostasis-related traits, respectively.
Footnotes
Acknowledgments
This study is part of the requirements for BP's doctoral degree in Biotechnology. The financial support for Agri-Bioinformatics Promotion Program provided by Bioinformatics Initiative Division, Department of Information Technology, Ministry of Communications and Information Technology, Government of India, New Delhi is gratefully acknowledged.
Author Contributions
Conceived and designed the experiments: PS, BP. Analyzed the data: BP. Wrote the first draft of the manuscript: BP, PS. Contributed to the writing of the manuscript: BP, PS. Agree with manuscript results and conclusions: BP, DMP, PS, RC, IS. Jointly developed the structure and arguments for the paper: BP, PS. Made critical revisions and approved final version: BP, PS, DMP, RC, IS. All authors reviewed and approved of the final manuscript.
Funding
This work was partially funded by Indian Council of Agricultural Research (ICAR) Grant-in-Aid DWR/RP/2010-5.3 to PS and support from Agri-Bioinformatics Promotion Program of Ministry of Communications and Information Technology, New Delhi to RC.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.