
Abstract
Phylogenetic recombination detection is a fundamental task in bioinformatics and evolutionary biology. Most of the computational tools developed to attack this important problem are not integrated into the growing suite of R packages for statistical analysis of molecular sequences. Here, we present an R package, rbrothers, that makes a Bayesian multiple change-point model, one of the most sophisticated model-based phylogenetic recombination tools, available to R users. Moreover, we equip the Bayesian change-point model with a set of pre- and post- processing routines that will broaden the application domain of this recombination detection framework. Specifically, we implement an algorithm that forms the set of input trees required by multiple change-point models. We also provide functionality for checking Markov chain Monte Carlo convergence and creating estimation result summaries and graphics. Using rbrothers, we perform a comparative analysis of two Salmonella enterica genes, fimA and fimH, that encode major and adhesive subunits of the type 1 fimbriae, respectively. We believe that rbrothers, available at R-Forge: http://evolmod.r-forge.r-project.org/, will allow researchers to incorporate recombination detection into phylogenetic workflows already implemented in R.
Introduction
Recombination is one of the main mechanisms generating genetic variation. Failure to properly account for recombination can seriously undermine the validity of molecular evolution studies. 1 The need to account for recombination led to the development of a number of recombination detection programs (see http://bioinf.man.ac.uk/recombination/ for a comprehensive list). Recombination detection algorithms can be separated into 4 main categories: distance-based, phylogenetic-based, compatibility-based, and substitution distribution-based. 2 Here, we concentrate on phylogenetic-based methods that detect discordant phylogenetic relationships along a sequence alignment. The most sophisticated model-based recombination detection methods rely either on hidden Markov models 3 or on Bayesian multiple change-point models. 4 We chose to work with the Bayesian dual multiple change-point (DMCP) model of Minin et al, 5 implemented in the Java package DualBrothers, because this approach has been successfully used in a wide range of molecular evolution studies that include detecting recombination in rhodopsin genes in freshwater bacterioplankton, 6 studying lateral gene transfer in prokaryotes, 7 and selecting non-recombinant portions of genes for genealogical reconstruction in Leavenworthia alabamica, 8 to name a few. Moreover, DualBrothers proved to be more accurate in estimating recombination breakpoint locations relative to other competing approaches. 2
Software Description
The Bayesian DMCP model detects recombination while accounting for changes both in tree topology and evolutionary rates across the nucleotides of aligned DNA sequences. The posterior distribution of the DMCP model is analytically intractable, so the model parameters are approximated by a Markov chain Monte Carlo (MCMC) algorithm. As with many Bayesian phylogenetic methods, the most challenging aspect of the MCMC in the context of the DMCP model is sampling over tree topologies that relate molecular sequences under study. When the number of sequences exceeds 6, it becomes computationally infeasible to explore all of the possible topologies during MCMC. To address this issue, DualBrothers restricts the search of the tree space to a pre-specified set of topologies. To produce a reasonable set of candidate trees we use a sliding window approach, repeatedly restricting attention to a subset of the alignment sites. 9 Our package, rbrothers, uses BIONJ, a neighbor joining tree reconstruction algorithm, 10 to create a candidate tree from each sliding window that we shift along the alignment by a pre-specified number of sites. The BIONJ-based phylogenetic reconstruction is repeated until we reach the end of the alignment. For some data, each sliding window may not contain enough information to reconstruct a phylogeny with confidence, leading to omission of highly probable trees during the formation of the set of candidate trees. To overcome this difficulty, rbrothers includes a novel bootstrapping option, which instructs rbrothers to create a bootstrap sample of phylogenies estimated by the BIONJ method for each sliding window.
As input, rbrothers takes aligned sequences in either Phylip or Fasta format. If the alignment contains less than 7 sequences, all possible unrooted phylogenetic topologies will be considered during the DMCP model-based recombination detection. If there are more than 6 sequences, a window size and a step size are required in order to form a set of candidate trees. After pre-prosessing steps that heavily rely on the R package ape, 11 rbrothers calls DualBrothers via the rJava package to produce a MCMC sample approximating the posterior distribution of all model parameters. To monitor convergence and mixing of MCMC, rbrothers uses the coda package 12 and provides a trace plot of the log likelihood along with an autocorrelation plot of the log likelihood via a single command. Our package, rbrothers, makes use of rJava to interface with DualBrothers, allowing the package to be used on all major operating system platforms (Windows, Mac OSX, and Linux). The rbrothers source code and a companion web page (http://evolmod.r-forge.r-project.org/) contain extensive documentation, a detailed tutorial reproducing 1 of the examples from Minin et al, 5 and 2 demo R scripts that can be run in R with the help of the demo() command. To demonstrate the ease of use of rbrothers, we present an example of a novel recombination analysis of 2 Salmonella fimbrial genes.
Two Salmonella Fimbrial Genes
Salmonella enterica subspecies enterica–-a subspecies of Salmonella enterica–-contains a majority of the strains pathogenic to humans. 13 Uncovering mechanisms of pathadaptive evolution of these strains is important for understanding Salmonella enterica pathogenicity. A recent study demonstrated that point substitutions in a gene coding for the type 1 fimbrial adhesin, FimH, exhibit signatures of recent positive selection. 14 However, it is likely that the evolution of fimH gene is shaped by both point substitutions and intragenic recombination. Therefore, we investigate the presence of recombination in the fimH gene. For comparison, we also examine fimA that encodes the major structural subunit of the Salmonella type 1 fimbriae.
We start with fimH and fimA sequences from 8 Salmonella strains representing 8 serovars: Typhimurium strain LT2 (TmLT 2), Paratyphi A strain ATCC 9150 (ParaA 9150), Paratyphi B strain SPB7 (ParaBSPB 7), Paratyphi C strain RKS4594 (ParaC 4594), Typhi strain CT18 (TyphiCT 18), Gallinarum strain 287/91 (Galli 28791), Newport strain SL254 (NewportSL 254), and Kentucky strain CVM29188 (Kentuc 29188). Since sequence divergence is very low in these 2 alignments (average pair-wise sequence diversity π being 0.020 ± 0.003 for fimH and 0.015 ± 0.004 for fimA), we set prior odds of at least 1 break-point to 1:1000 to avoid inferring spurious recombination events. Notice that this is in contrast to the default 1:1 odds in DualBrothers. We choose to work with the low prior odds of at least 1 breakpoint because the posterior distribution of the number of break-points was highly sensitive to the prior. The default DualBrothers's prior results in a large number of estimated recombination break-points, most of which are likely spurious. Gradually decreasing the prior mean of the number of break-points corresponds to a gradual decrease in the number of estimated break-points, but after a certain point the posterior stops being sensitive to the prior, strongly supporting 2 break-points. When we report only these 2 strongly supported break-points we are being very conservative in the sense that there are probably more recombination break-points supported by these data. However, interpreting the posterior distribution of break-points with a more liberal prior on their average proved to be challenging. The above analysis underscores the importance of prior sensitivity analysis when performing Bayesian inference with complex models.
MCMC for the fimH alignment mixed with less success than MCMC for the fimA data, prompting us to run the former Markov chain for 51 × 10
6
iterations, discarding the first 10
6
iterations, and the latter chain for 2.1 × 10
6
iterations, discarding the first 10
5
iterations. The latter is the default setting in rbrothers. In the fimH alignment, the DMCP model produces strong evidence for at least two recombination break-points at sites 412 and 591. The corresponding Bayesian credible intervals, computed according to the procedure described in Minin et al,
5
are (392, 485) and (585, 596). In contrast to the fimH analysis, rbrothers finds no sign of recombination in the fimA alignment. In Figure 1, we plot the posterior probabilities of the phylogenetic tree topologies for each site in the 2 alignments, with corresponding trees shown below the probability plots. We only plot trees whose posterior probabilities were above 0.5 at some sites of the sequence alignment. The above results can be reproduced by running a corresponding demo example in the rbrothers package:

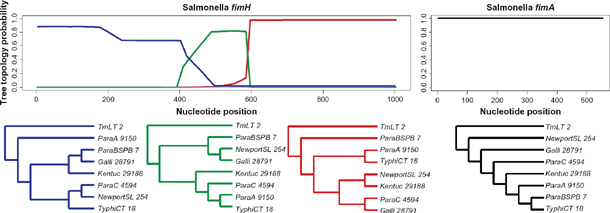
The top plots show site-specific posterior probabilities for the top 3 most probable phylogenetic tree topologies in the fimH alignment and for the most probable tree in the fimA alignment. Phylograms corresponding to the 4 unrooted tree topologies are shown underneath.
Using rbrothers within the R Package Ecosystem
One of the advantages of having a DMCP model implementation available in R is the possibility of combining the results of this inferential framework with other phylogenetic analyses. In fact, rbrothers already uses ape's excellent graphical facilities to display DMCP results seamlessly and intuitively. 12 When the sequence alignment contains a small number of well estimated break-points, the DMCP framework provides an estimated segmentation of the alignment into blocks, each supporting a different phylogenetic tree, as shown in the Salmonella example above. This segmentation can be used further to map mutations onto corresponding phylogenies or to test hypotheses about relatedness of the sequences under study using existing R packages, such as ape and phangorn.11,15 A more rigorous approach, in which the downstream analyses would be integrated over the posterior distribution of alignment segmentations, is also possible and will be subject to future research and software development.
Funding
This work was supported by the National Science Foundation [DMS-0856099]; and the National Institutes of Health [1RC4AI092828-01, R01 GM084318].
Footnotes
Author Contributions
Conceived and designed the experiments: JI, SC, EVS, VNM. Analyzed the data: JI, SC. Wrote the first draft of the manuscript: JI, VNM. Contributed to the writing of the manuscript: JI, SC, EVS, VNM. Agree with manuscript results and conclusions: JI, SC, EVS, VNM. Jointly developed the structure and arguments for the paper: JI, SC, EVS, VNM. Made critical revisions and approved final version: JI, SC, EVS, VNM. All authors reviewed and approved of the final manuscript.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.