
Abstract
Hsp90 chaperone has been identified as an attractive pharmacological target to combat cancer. However, some metastatic tumors either fail to respond to Hsp90 inhibition or show recovery necessitating irreversible therapeutic strategies. In response to this enforced senescence has been proposed as an alternate strategy. Here, we demonstrate that inhibiting Hsp90 with 17AAG sensitizes human neuroblastoma to DNA damage response mediated cellular senescence. Among individual and combination drug treatments, 17AAG pre-treatment followed by doxorubicin treatment exhibited senescence-like characteristics such as increased nucleus to cytoplasm ratio, cell cycle arrest, SA-
Introduction
Heat shock proteins (Hsps) are implicated in evading stress induced cell death programming and in adaptive cellular responses to stress. 1 The Hsp90 chaperone functions are entangled with tumor progression identifying this protein as a cancer chaperone. 2 In support of this, the surface expression of Hsp90 in cancers of the human nervous system (NB69, SH-SY5Y, SK-N-SH) correlated with enhanced survival and proliferation, whose functional blockade with 17-allylamino-17-demethoxygeldanamycin (17AAG) or geldanamycin compromised cell survival.3–5 As an exception, the drug resistant and metastatic human neuroblastoma, IMR-32 failed to respond to 17AAG treatment 6 despite Hsp90 being the central regulator of cell signaling.7,8 The non-responsiveness may relate to major regulatory mechanisms such as tumor suppression.
In addition to essential oncogenic kinase activation, inactivation of tumor suppressor mechanisms involving p53-p21CIP/WAF-1 and pRb-p16INK4a are significantly implicated in cancer progression. Despite occurrence of mutations in p53 tumor suppressor gene in a large number of human tumors, neuroblastoma retains functional protein, and this could be the reason why inactivating mutations in p53 enhances chemotherapy induced cytotoxicity. 9 The neuroblastoma also comprise constitutive p16INK4a expression independent of Rb phosphorylation. 10 The constitutive p16INK4a expression was presumed to act against not only cytotoxic pathways but to bypass senescence programming. To circumvent chemotherapeutics induced non-selective cytotoxicity and to promote the therapeutic potential of pharmacological inhibitors of Hsp90 in metastatic tumors, several combination drug treatment strategies have been proposed.6–8 These combination treatments, however, suffer due to induced stress response mechanism, which may help in cell recovery and in making aggressive cancer phenotypes or affect bystander cells due to lack of tumor selectivity.
The DNA topoisomerase I inhibitor, doxorubicin has been shown to induce senescence-like phenotype in cells exposed to prolonged treatments. Subsequent to these findings, doxorubicin combination with Akt targeting was shown to be effective in Akt positive cells for cancer treatment. 11 Nevertheless, neuroblastoma cells possess altered PI3K-Akt-mTOR pathway activation that correlates with poor prognosis 12 and do not respond to Akt destabilization even with 17AAG induced disassociation of Akt from Hsp90 binding. 6 Considering the constraints associated with 17AAG response in IMR-32 neuroblastoma, we hypothesized that functional bereavement of Hsp90 status combining with doxorubicin may effectively combat cancer.
In the present study, we investigated the acceleration of pre-sensitizing effects of Hsp90 inhibition on doxorubicin induced cellular senescence. The acquired senescence phenotype was characterized by increased senescence associated
Materials and Methods
Cell Cultures, Treatments and Reagents
IMR-32, SRA01, and jurkat cells were from American Type Cell Culture. Cells were grown and maintained in DMEM containing 10% fetal bovine serum albumin in the presence of penicillin (100 U/mL), streptomycin (50 μg/mL) and kanamycin (30 μg/mL) at 37 °C in a humidified incubator with 5% CO2 supply. For treatments, 2 × 105 cells were grown on cover glass (22 × 22 mm, Fisher Scientific, USA) in a 6-well culture dish (nunc, Thermo Scientific, USA) and incubated in complete medium either with doxorubicin (Sigma-Aldrich, USA) or with 17AAG (Invivogen, USA) at different time intervals. The effective drug concentrations were standardized by analyzing their ability to induce cytotoxicity in case of doxorubicin and to degrade 90% of Hsp90 client proteins in case of 17AAG. The concentration used throughout the manuscript for doxorubicin was 0.1 μM and for 17AAG it was 2 μM.
Cell based assays
Fluorescence activated cell sorting (FACS, FACSCalibur, Becton Dickinson, USA) analysis for DNA content was performed by staining cells with propidium iodide (50 μg/mL; Sigma-Aldrich, USA). To measure intracellular reactive oxygen species, [(ROS)i], cells were stained with CM-H2DCFDA (5 μM; Dojindo, Japan), to measure intracellular calcium, [(Ca2+)i], cells were stained with Fluo 3-AM (1 μM; Dojindo, Japan), to measure a change in mitochondrial membrane potential (Δψ
Senescence associated β-galactosidase (SA-β-gal) staining
The phase-contrast images of cells were captured using Axiovert 200 microscope (20× magnification; Carl Zeiss, Germany). The SA-
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
The RNA was isolated using TRIZOL (Sigma-Aldrich, USA) method. The first strand cDNA was prepared from 1 μg total RNA using Prime Script 1st strand cDNA synthesis kit (Takara Bio Inc., Japan). The gene specific cDNA amplifications were performed using qualitative PCR primers in a dual block PCR machine (DNA Engine; Bio-Rad, USA). The primers used were cell cycle regulators:
Immunoblot analyses
Cell lysates were prepared using HEPES lysis buffer (20 mM HEPES, 10 mM NaCl, 1.5 mM MgCl2, 0.1% Triton X-100, pH 7.6), 20 μg total protein was run on 10% SDS-PAGE and was transferred on to nitrocellulose membrane. The primary antibodies, HRPO- and FITC-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology Inc., (USA).
Laser Scanning Confocal Imaging Microscopy
Staining for mitochondria and actin was performed in cells with CMX-
Rhodamine 123 (RH123) efflux assay
Cells were incubated with Rh123 (1 μM; Dojindo, Japan) and analyzed in the FACSCalibur. The Rh123 efflux ratio was calculated by dividing the mean channel number with cyclosporin A (CsA) and mean channel number with Rh123 alone.
Real-time polymerase chain reaction (real-time PCR)
The telomerase activity was measured by quantitative telomerase detection kit (US Biomax, USA). A standard real time PCR was run in Realplex Realtime PCR machine (Eppendorf Mastercycler ep gradient S, Germany) with the TSR oligonucleotide and the telomerase activity was calculated from the standard curve.
Colony forming assay (CFA)
Cells were mixed with molten soft agar at 37 °C, poured over a base layer of agar and allowed to grow in complete medium with 5% CO2 supply. After eight days, cells were stained with 0.1% crystal violet and observed under Axiovert 200 microscope in differential interference contrast microscope (DIC, 5× magnification). The colony size in micro meters was calculated from πr 2 and plotted.
Neo-vascularization assay
Cover glasses were pre-coated with matrigel (BD Biosciences, USA) for 45 min and cells were spread on matrigel, incubated with complete medium containing the drugs for 24 h and the tube or colony formation was observed under Axiovert 200 microscope in DIC (5× magnification).
siRNA Knockdown of Hsp90
Hsp90 siRNA was designed using Invitrogen BLOCK-iT™ RNAi designer software from HSP90 cDNA (Accession No. NM_005348.2). The three siRNA used in the present study were, oligo1, 5′-GAA CAAA CAAGATCGAACTCT-3′; oligo2, 5′-GAGA GAGCT CATTTCAAATTCATCA-3′; oligo3, 5′-ACTCTGG GAAAGAGCTGCATATTAA-3′. The siRNA was introduced into the cells using nanoparticle based X-fect transfection reagent (Clontech, USA).
Evaluation of conditioned medium (CM) for senescence promoting secretory factors (SASPs)
IMR-32 cells were 17AAG pre-treated for 24 h followed by doxorubicin for 5 days, and after confirming the SA-
Statistical analyses
The data presented are mean ± SEM of minimum three independent experiments. The significance was calculated by student's
Results
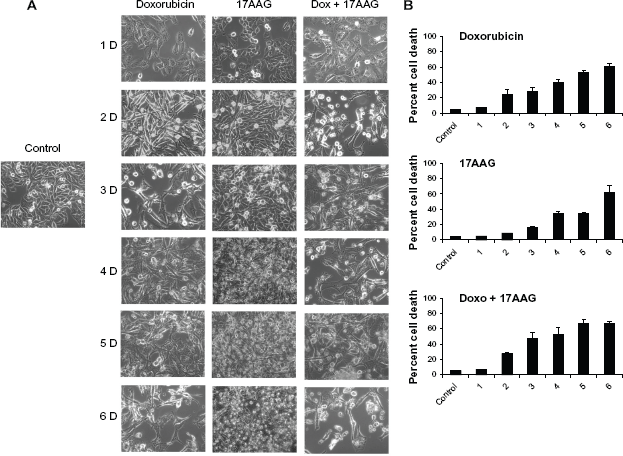
17AAG combination decreases doxorubicin induced senescence response
Senescent cell morphology is typically associated with increased nucleus to cytoplasm ratio with protracted cellular extensions and increased SA-
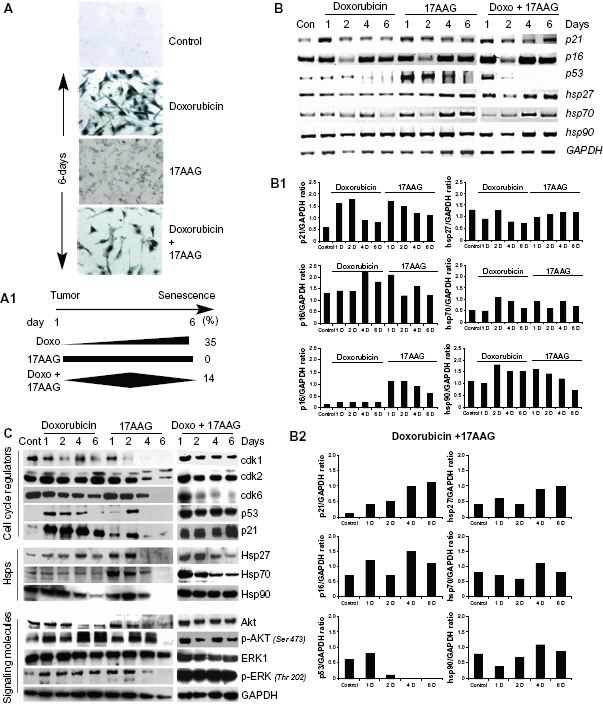
Effect of doxorubicin, 17AAG and their combination treatments on IMR-32 neuroblastoma cells.
To understand the indistinctness in
17AAG and its combination with doxorubicin induces stress response
Tumor suppressors play a major role in deciding the fate of cells under stress conditions. To investigate their functional role in senescence, the expression levels of p21CIP/WAF-1, p16INK4a and p53 were examined by RT-PCR analyses at 2-, 4- and 6-day post-treatments. The individual treatments showed a gradual decrease in p21CIP/WAF-1 along with the time of treatment. The combination treatment though showed an initial decrease in 2-day treatment, and a significant increase was observed by a 6-day treatment. Correlating with p21CIP/WAF-1 there was an increase in p53 expression in 17AAG (up to 4-day) and its combination with doxorubicin (by 1-day) corresponding to activation of cellular stress response. The doxorubicin induced p53, however, may be related to DNA damage response. Additionally, increased expression of stress proteins, Hsp27, Hsp70 and Hsp90 upon 17AAG and its combination with doxorubicin treatments further confirm activation of stress response (Fig. 1B). The ratio between each target gene and GAPDH expression was calculated using ‘Image J’ analysis to normalize loading controls and represented in bar diagram. Therefore the bar diagrams represent fold gene expression compared to GAPDH (Fig. 1B1 and Fig. 1B2).
To study protein levels of tumor suppressors, signal transduction and stress proteins, cell lysates collected at day 2, 4 and 6 of the treatments were examined by immunoblot analysis. Prolonged treatment of cells with individual drugs displayed gradual but radical decrease in cyclin dependent kinases, cdk1, cdk2 and cdk6, which has implications both in G1/S and G2/M cell cycle transitions. Prolonged combination treatment displayed a decrease in cdk6 alone that is involved in the G0 to G1 cell cycle transition. Doxorubicin and 17AAG treatments showed decrease in Hsp27, Hsp70 and Hsp90 levels; interestingly, only Hsp90 levels were unaffected by the combination treatment. Consistent with Hsp90 levels, Akt and ERK1/2 basal levels were not affected by the combination treatment compared to individual treatments. However, the phosphorylation status of Akt and ERK1/2 was found to be decreased (Fig. 1C).
17AAG pre-treatment but not the combination treatment accelerates senescence response
We excluded the combination treatments in our subsequent study, because prolonged treatments interfered with senescence activation by inducing early cytotoxicity (Suppl. Fig. 1B). In the following experiments, we studied pre-sensitizing effects of drugs on senescence acceleration. In brief, cells were treated with doxorubicin or 17AAG for 24 h and the drug containing medium was replaced with fresh medium containing 17AAG or doxorubicin for 5-days were examined for SA-
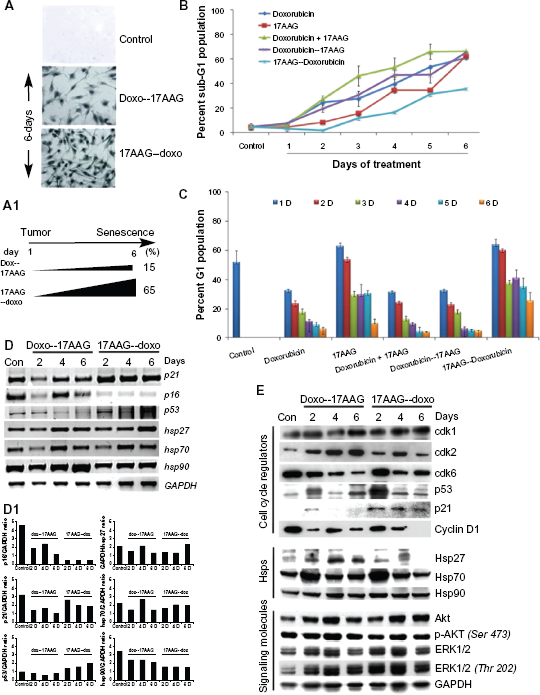
Effect of pre-treatments on IMR-32 neuroblastoma cells.
17AAG pre-treatment retains survival potential
To investigate the difference in molecular mechanism of senescence activation in the pre-treatments, the RT-PCR analysis of cell cycle regulators was performed. A gradual decrease in p21CIP/WAF-1 in doxorubicin pre-treatment and a steady increase in 17AAG pre-treatment correlated with increased expression of p53 suggesting involvement of p53 and p21CIP/WAF-1 in senescence activation. The increased p21CIP/WAF-1 but not p16INK4a expression correlated with enhanced SA-
Next, we examined protein profiles of regulatory molecules by immunoblot analysis using lysates of drug treated cells at days 2, 4 and 6. Though there was some fluctuation in cdk1 levels, no significant change was observed both in doxorubicin pre-treatment or 17AAG pre-treatments suggesting no effect on mitosis. Increased cdk2 in doxorubicin pre-treatment suggested G1 clearing of cells but variability in 17AAG pre-treatment suggest crisis in cellular decision making. The decrease in cdk6 levels in both the pre-treatments suggests inability of cells to re-enter cell cycle and can be argued with irreversible effects of cell cycle inhibition. While Hsp90 levels were unaffected by neither of pre-treatments, Hsp27 and Hsp70 levels showed an initial increase in a 2-day treatment but its normalization by 6-day treatment. Correlating with increased levels of p53 and p21CIP/WAF-1, a decrease in cyclin D1 levels suggested decreased proliferation potential. In contrast to individual and combination treatments, in the prtreatments, a chronic activation of Akt and ERK1/2 kinases suggested maintenance of survival potential (Fig. 2E). Considering the accelerated senescence with 17AAG pre-treatment with reduced cytotoxicity, the subsequent experiments were performed with 17AAG pre-treatments.
17AAG pre-treatment shows typical features of senescence
Actin reorganization and nuclear accumulation was proposed as an additional marker for cellular senescence.
15
To understand cellular actin dynamics, actin organization was examined in 17AAG pretreated senescent cells with Oregon Green Phalloidin. Compared to untreated cells, senescent cells showed a time dependent increase in peripheral actin bundling and accumulation of non-polymerized actin in the nucleus. Since sustained mitochondrial functions promote senescence activation,
16
mitochondria organization was also examined with mitotracker red (CMX-
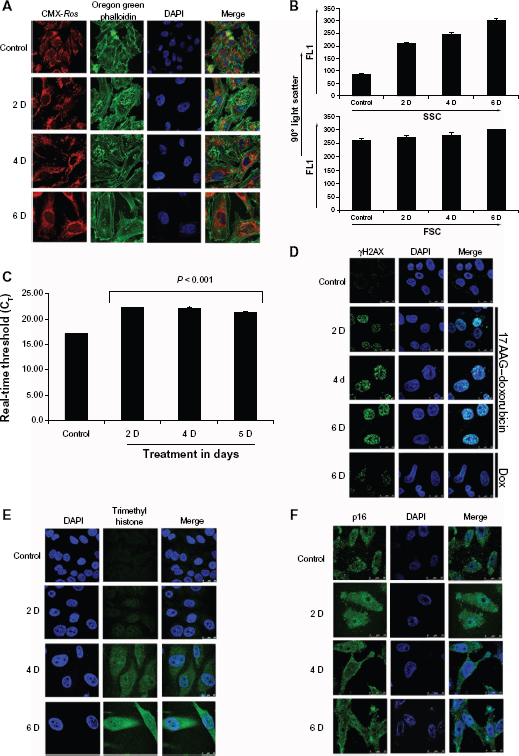
Molecular characterization of 17AAG pre-treatment accelerated cellular senescence.
Accumulation of cells in G1 though suggested replicative senescence, involvement of p53- p21CIP/WAF-1 pathway highlighted stress induced senescence. To further distinguish the mode of senescence on 17AAG pre-treatment, telomerase activity was measured from senescent cells. A small increase in telomerase activity and its stabilization after 2-day treatment suggested cellular attempt to bypass senescence reprogramming (Fig. 3C), which is a characteristic feature of cancer cells. 18 While H3K4me3 marks transcription start sites of active genes, 19 senescence associated heterochromatin foci (SAHF) marks DNA damage response (DDR) to senescence. 20 The cytoimmunofluorescence analyses of γH2AX (for SAHF) and H3K4me3 showed a gradual increase in SAHF but cytosolic accumulation of H3K4me3 (Fig. 3D and E respectively). Telomerase activity is linked to p16INK4a, 21 however, neuroblastoma exhibit chronic p16INK4a expression (Fig. 1B), which is thought to bypass senescence programming. 22 To examine p16INK4a cellular distribution, we performed cytoimmunoflourescence. Although, there was an increase in nuclear distribution in 2-day treatment, which has significantly decreased in 4- and 6-day treatments invalidating its role in senescence activation (Fig. 3F).
17AAG pre-treatment induced [(ROS)i] and [(Ca2+)i] promotes senescence and autophagy
Calcium permeation across membranes and mobilization from organelles was associated with senescence
23
along with increased [(ROS)i].
24
We measured [(Ca2+)i] and [(ROS)i] levels by FACS. In comparison with the control, we observed an initial increase in [(Ca2+)i] levels by 24.7 folds in 2-day treatment, which was decreased by 17.3 folds and 18.1 folds by day-4 and day-6 treatments (Fig. 4A,
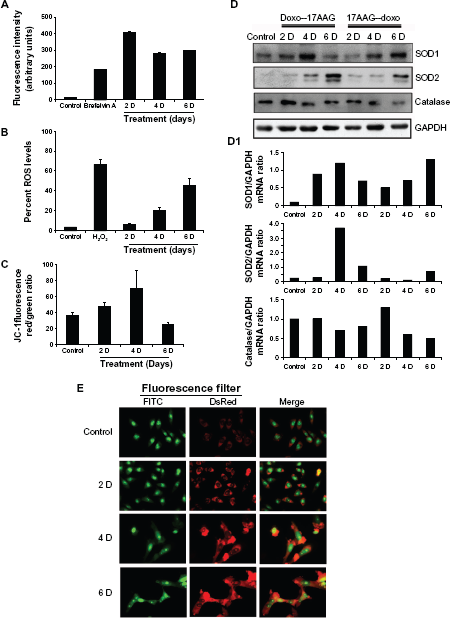
Effects of 17AAG pre-treatment on cellular redox status.
Since oxidative stress is caused by the imbalance between [(ROS)i] and impairment of antioxidative enzyme system, 26 we examined the status of antioxidative enzymes, superoxide dismutases, SOD1 (CuZn SOD, cytoplasmic), SOD2 (MnSOD, mitochondrial) and catalase (peroxisomes) in both doxorubicin and 17AAG pre-treatments by immunoblot analysis. The induced levels of SOD1, SOD2 and decreased levels of catalase on both the pre-treatments suggested activation of intracellular antioxidative defense, which also advocates non-lethal level production of [(ROS)i] (Fig. 4D). The gene expression analyzed by Image J and represented as gene versus GAPDH ratio (Fig. 4D1). In agreement with moderate increases in oxidative stress that correlated to sensitizing effects of cellular senescence 27 but not to apoptosis, we also observed a moderate increase in [(ROS)i]. Since senescent cells are usually resistant to apoptosis or necrosis, removal of damaged cells could occur by autophagy (type II programmed cell death). Accordingly, senescent cells showed enhanced acridine orange staining, a characteristic feature of macroautophagy 14 (Fig. 4E).
17AAG pre-treatment induced senescence is irreversible
Pharmacological modulator curcumin was shown to promote proliferation in neuroblastoma,
6
therefore, we examined the effect of curcumin on reversal of senescence. Although curcumin (15 μM, 24 h) increased the number of SA-
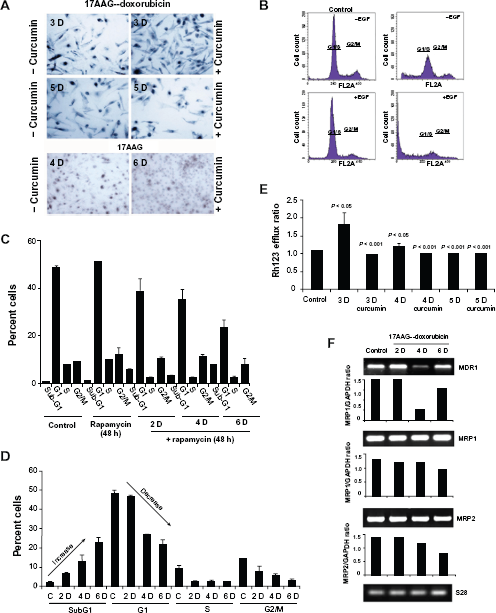
Studies on 17AAG pre-treatment accelerated senescence reversal.
The mTOR, a serine/threonine kinase activation favors senescence in tumor cells.
28
To investigate the involvement of mTOR signaling in senescence, cells were treated with mTOR inhibitor, rapamycin for 48 h and compared with rapamycin (200 nM) combined 17AAG pre-treatments at day 2, 4, and 6 using FACS. While rapamycin had no effect on control cells, its treatment to senescence directed cells resulted in a decrease to G1 and increase to subG1 population suggesting that compromised mTOR functions compromises cell survival (Fig. 5C,
Enhanced multidrug resistance (MDR) is one of the characteristic features of senescent cells.
29
To understand whether senescent cells show any enhanced drug resistance, we examined for Rh123 efflux of cells from day 3, 4 and 5 of the treatment. Since curcumin interferes with MDR functions, curcumin was used (2 h pre-treatment prior to Rh123 efflux assay) to inhibit MDR functions. Compared to control efflux ratio, which was due to constitutive MDR activation in IMR-32 (9), 3-day treatments showed increased efflux ratio by 0.5 folds, suggesting enhanced functions of MDR on the onset of senescence. Further, curcumin was effective in decreasing MDR functions as observed by Rh123 efflux (Fig. 5E). Concurrently, decreased
Senescent cells show compromised colony forming ability and neo-vascularization
It was reported that only a subset of cancer cells respond to drug induced cellular senescence and remaining cells retain metastatic potential and lead to recurrence.30,31 To exclude the possibility of tumor relapse in our senescence model, first we examined anchorage-independent cell growth using soft agar colony formation assay. Cells from post 17AAG pre-treatments at day 2, 4 and 6 were placed on agarose coated surface and the proliferation potential of cells was monitored for colony forming ability. Cells collected on the onset of senescence showed ±65% decrease in colony size (Fig. 6A and 6A1). Next, we examined for neo-vascularization after growing cells on matrigel coated surface. A 6-day growth of control cells on matrigel resulted in organized large colonies. However, cells from the onset of senescence showed a significant decrease in the organized colony structures (Fig. 6B). These findings are consistent with SA-
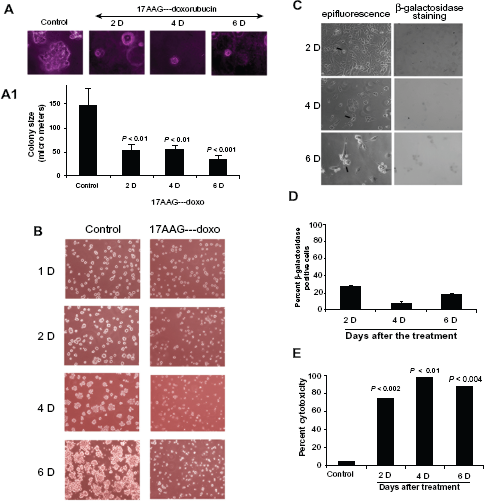
Anti-proliferative/anti-neo-vascularization effects of 17AAG pre-treatment accelerated senescence cells.
Compromising Hsp90 expression compromises survival potential of senescent cells
We show that interference with high affinity conformation of Hsp90 with 17AAG
32
accelerates doxorubicin induced senescence. In these experiments 17AAG treatment
CM-SASPs from senescent cells induce senescence
Enforced senescence in tumor cells has also been proposed to be deleterious, because it activates age-associated pathologies.
34
The genotoxic stress enhances senescence associated secretory phenotype (SASP), which however, is linked to the loss of p53 function or enhanced oncogene activation.
35
Since, IMR-32 exhibit functional p53 and chronic oncogene activation, we examined CM containing SASPs from senescent cells on pro-senescence activity. Between the two cell types used in our study, jurkat cells mimic T-cell leukemia and secret cytokines, whereas, SRA01 are virus free immortalized cells containing progenitors of myoepithelial lacking tumorigenic activity. The CM treated tumor cells showed senescence morphology by 4-day treatment as observed by SA-
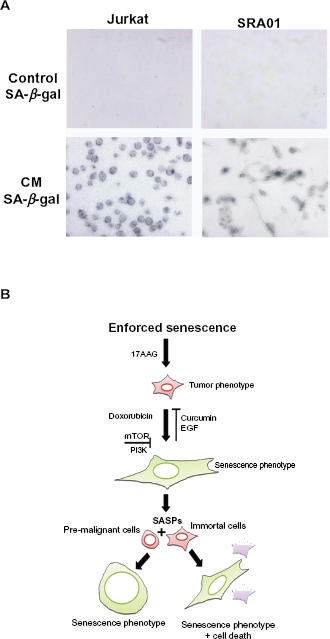
Discussion
Neuroblastoma has been considered as a malignant manifestation of aberrant sympathetic nervous system development. Irreconcilable differences in therapeutic strategies due to high tumor heterogeneity limit existing approaches, thus, insist on developing novel strategies, which can exploit the oncogenic signatures of tumor cells. 36 Earlier, we showed that pharmacological targeting of Hsp90 functions deprives proliferation promoting signal transduction in IMR-32 neuroblastoma, however, the apparent recurrence on post-treatment suggested limitations to this approach.6,37 In this study, we demonstrate how compromising the chaperoning functions of Hsp90 (17AAG pre-treatment) sensitizes neuroblastoma to doxorubicin induced cellular senescence, and present conformation specific Hsp90 as a barrier to enforced senescence of tumor cells.
Although Hsp90 is an exciting drug target due to its cancer specificity it is likely to induce drug resistance over time.
38
Some Hsp90 inhibitors can induce oxidative stress leading to non-specific actions in tumor cells and may also affect bystander cells.
39
However, in the present study, microgram concentrations of 17AAG for prolonged incubation periods is required to obtain 50% cells death, which may develop drug resistance in cancer cells in due course of time. Similar findings with doxorubicin suggested ability of cells to resist chemotherapeutic intervention. Although combination drug treatment has improved the cytotoxic effects of drugs in earlier time periods, prolonged periods of incubation suggested either delayed drug response or acquired drug resistance (Supplemental Fig. 1B). Doxorubicin pre-treatment followed by 17AAG did not improve efficacy of cytotoxic treatment strategy, but 17AAG-pretreatment has significantly decreased cytotoxicity (Supplemental Fig. 2B). To our surprise, SA-
In recent years, enforcing premature senescence in tumor cells with chemotherapeutic mediation has been proposed as an alternate strategy to combat cancers.40,41 Prolonged cytostasis either through p53-p21CIP/WAF-1 or pRb-p16INK4a meditation was thought to be the focal point in such models. 42 Despite the fact that individual drug treatments do not show significant increase in p21CIP/WAF-1 expression, the combination treatment showed increased p21CIP/WAF-1 expression correlating with p53 (compare Fig. 1B and C with Fig. 2D and E) that however, did not result in increased senescence positive cells (compare Fig. 1A1 with Fig. 2A1). The increased p21CIP/WAF-1 expression only in 17AAG pre-treatment but not in doxorubicin pre-treatment resulted in increased senescence positive cells (Fig. 2). Although, replicative senescence is linked to p16INK4a, in our study, incessant p16INK4a over expression was found to have no influence on induced senescence signaling in individual and combination drug treatments (Fig. 1), but its decrease in 17AAG pre-treatment resulted in significant increase in senescence positive cells (Fig. 2). We speculate that p16INK4a may be antagonizing senescence signaling induced by chemotherapeutic interventions. In accordance with this, p16INK4a constitutive expression 22 has been implicated in bypassing cellular senescence. 43 We are in the process of examining the role of p16INK4a in tumor metastasis and bypassing induced senescence. Excluding p16INK4a in 17AAG pre-treatment induced senescence, the senescence observed in our model is pragmatic to p53-p21CIP/WAF-1 mediation.
Interestingly, the stress proteins (Hsp27, Hsp70 and Hsp90) that showed fluctuations in individual and combination drug treatments (Fig. 1) were not significantly affected by the followed drug treatments (Fig. 2) implicating functional compromise but not expression levels play role in altering the cell fate. Consistent with this stabilized p53 and p21CIP/WAF-1 levels appeared to play significant role in cellular decision correlating with differentially regulated cdks. In accordance with this we did not find complete loss of proliferation (PI3K-AKT) or survival (Ras-Raf-ERK) signal transduction but their apparent alteration (Fig. 1 and Fig. 2). The continued kinase activities may therefore relate to cellular crisis induced by the combination drug treatments. The data obtained from Figures 1 and 2 however failed to conform to any classical signal transduction pathway leading to senescence, it confirmed that senescence occurred through p53-p21CIP/WAF-1 mediation. With this lead we went ahead for further characterization of senescence.
Enhanced SAHF is involved in maintaining the senescence phenotype. Although SA-
Physiological factors such as ROS and Ca2+ were also implicated in senescence signaling. In fact aging phenotypes were thought to be established through oxidative signaling. Since Hsp90 inhibitors alone can elevate ROS, 39 in the present study, the ROS mediated senescence activation48,49 cannot be exempted. To our surprise, we observed only a small but gradual increase in ROS levels, which were addressed by the activated mitochondrial antioxidant defense. These findings sets aside the role of ROS in senescence activation.27,50,51 Although Hsp90 inhibition induces the release of intracellular Ca2+, 52 in a rat tumor model, this release was then correlated to decreased functions of Hsps. 53 Unlike the rat model where Hsp gene transcription was found to be compromised, IMR-32 cells retained some Hsps suggesting survival potential of cells upon elevated Ca2+ levels. Reinforcing this, the increased calcium levels correlated with the activation of senescence response but not apoptosis (Fig. 4). In previous sections we discussed how senescent cells are resistant to cell death mechanisms such as apoptosis under conditions of depleted oxygen stress 54 and now in agreement with this, in the present study, we observed activation of autophagy, which was considered to be pro-survival response mechanism.
Senescence mediated by p53-p21CIP/WAF-1 has been reported to be reversible therefore may limit the application of our strategy. Failing to induce senescence reversal with mTOR inhibitor, EGF or curcumin that were known to decelerates senescence,55,56 appraise that enforced proliferation stimulus is inoperative once senescence signal is being activated. In accordance with earlier understanding that synergistic activation of PI3K and mTOR provide survival fitness to cancer cells, 57 and their chronic activation lead to senescence, 58 we demonstrated that drug treatments promote senescence through chronic signal activation (Fig. 5). Therefore the irreversible potential of tumor cells may relate to enhanced senescence-associated multi drug resistance. 59 Doxorubicin being the substrate of multi drug resistance gene product, MDR1, 60 it was presumed that Hsp90 inhibition promotes its bioavailability to exhibit enhanced senescence activity. With the prelude that Hsps promote multidrug resistance in tumor cells, we interpret that functional Hsp90 inhibition in MDR1 positive 9 IMR-32 cells could possibly have primarily compromised the drug efflux, thus could have increased doxorubicin cellular accumulation to promote senescence associated effects.
The augmentation of doxorubicin induced senescence by Hsp90 inhibitors in neuroblastoma suggests an alternate strategy to combat cancers. Evaluation whether enforced cellular senescence act as a tumor suppression mechanism by anchorage independent (growth on agarose) and anchorage dependent (growth on matrigel) growth assays revealed anti-pro-liferative and anti-angiogenic competence. However, in contrast to compromising of Hsp90 chaperoning function using pharmacological drugs, knockdown of Hsp90 using siRNA has significantly compromised the survival potential of senescent population (Fig. 6) suggesting Hsp90 expression is indispensible for cell survival whether cells are from normal, tumor or senescent groups.
Paradoxically chemotherapeutic drugs must exert two important tumor suppressor mechanisms namely, senescence or apoptosis. Hsps antagonize apoptotic signaling1,53 but facilitates senescence. 61 The conformational maturation and functional stabilization of oncogenes was aided by conformation specific Hsp90 (high affinity conformation) present in tumor cells, which is in addition to the presence of normal Hsp90 (low affinity conformation). 32 In the present study, pre-sensitization of tumor cells with conformation specific anti-Hsp90 inhibitor, 17AAG to doxorubicin treatment suggested that by compromising high affinity conformation of Hsp90 it may be possible to sensitize Hsp90 inhibition resistant tumor cells to chemotherapeutic intervention. Our results demonstrate that Hsp90 in its high affinity conformation undeniably acts as an impediment to senescence signaling due to its involvement in tumor progression. Our findings are in agreement with earlier hypothesis that decreased chaperoning functions of Hsps promote senescence signaling.62,63
Findings from Campici's group 64 though projected enforced cellular senescence as a tumor suppressor mechanism, later findings from the same group showed that factors secreted to the culture medium (condition medium-CM) by senescent cells (SASPs) can promote transformation in pre-malignant cells.34,65 These findings invalidated the strategy of enforced cellular senescence for antitumor treatment. 66 Nevertheless, we project that CM from 17AAG pre-treatment induced senescent cells lack proliferation stimulus as envisioned by other groups but interestingly accelerated senescence-like phenotype in target cells (Fig. 7A). The disparity in SASPs from others to our study was found to be only the use of Hsp90 inhibition to accelerate senescence. Our findings, therefore, may have clinical benefits for cutting-edge treatments using anti-Hsp90 drugs. For thorough understanding of the molecular cross-talk in neuroblastoma, at this point in time we are characterizing SASPs and also investigating the functional silencing of p16INK4a and its contribution in bypassing senescence in IMR-32 cells.
In summary, both cancer and senescence cells express high amounts of Hsps, however, differ in their chaperoning activities. While enhanced chaperoning functions of Hsp90 with increased ATP binding affinity promote tumor progression allowing mutated gene products to function normally, the basal functions help to maintain protein homeostasis. Taking leads from these findings, we manipulated the chaperone functions of Hsp90 and studied genotoxic drug-induced cellular senescence in IMR-32 neuroblastoma. Among different individual and combination drug treatments, pre-sensitization of neuroblastoma with 17AAG accelerated doxorubicin induced DDR and signaled cells to stress induced cellular senescence. Despite its known functions in replicative senescence67,68 we demonstrate how DDR signaling promote stress-induced senescence. Several cellular factors, signaling molecules and second messengers that have contributed to enforced senescence in IMR-32 neuroblastoma were discussed. Essentially Hsp90 as a barrier for enforced senescence in tumor cells was emphasized (Fig. 7B). Our findings add contemporary information, attractive strategy and effective therapeutic option wherein exploitation of conformation specific Hsp90 in sensitizing metastatic tumor cells leads to enforced cellular senescence.
Author Contributions
US, KRP, JUK and VS performed the experiments and acquired the data. ASS conceived the hypothesis, designed the experiments, analyzed the data and wrote the manuscript.
Abbreviations
heat shock protein
90 kDa heat shock protein
17-allylamino-17-demethoxygeldanamycin
senescence-associated βgalactosidase
Trimethylation of histone H3 at lysine 4
DNA damage response
phosphorylated histone H2A
intracellular reactive oxygen species
intracellular calcium; Δψm change in mitochondrial membrane potential
conditioned medium
senescence-associated secretory phenotype
senescence associated heterochromatin foci
Rhodamine 123
fluorescence activated cell sorting
multidrug resistance gene
multidrug related protein
p21 cdk interacting protein 1/wild type p53 activated fragment 1
p16 inhibitors of CDK4
super oxide dismutase
small interfering RNA.
Funding
This work was supported by grants to ASS from Department of Science and Technology, Government of India.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Effect of 17AAG, doxorubicin and their combination on cell morphology and cytotoxicity. (A) Cells after respective treatments were observed under microscope and phase contrast images in 10× magnification were represented. (B) Cells were stained with propidium iodide and the DNA content was analyzed by FACS.
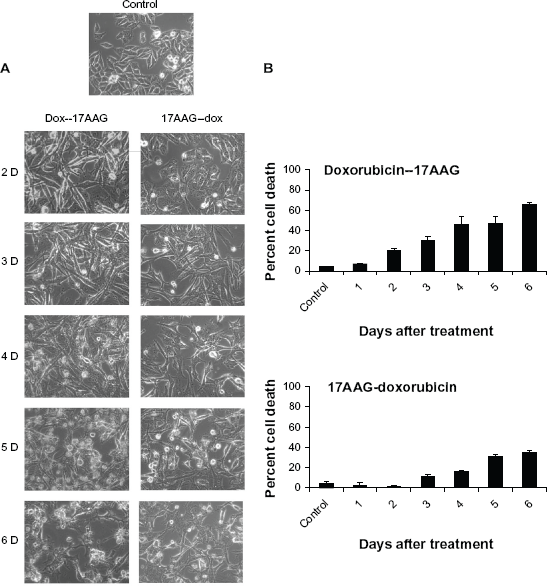
Effect of drugs pre-treatments on cell morphology and cytotoxicity. (A) Cells either treated with doxorubicin or 17AAG for 24 h followed by second drug treatment for 5-days were observed under microscope and the phase contrast images were represented. (B) Cells stained with propidium iodide and the DNA content was analyzed by FACS.
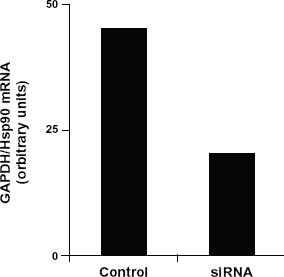
The RT-PCR analysis of siRNA to Hsp90 transfected cells.