
Abstract
Background
AlphaIIb/beta3 (αIIb/β3) complex is an important integrin that is involved in the final step of platelet aggregation. Peptides derived from either αIIb or β3 have demonstrated to have an effect on the activation of the complex and its ability to bind fibrinogen. We have previously defined a peptide from β3, which inhibits agonists-induced platelet aggregation.
Methods
We used standard methodologies for construction of clones and expression of cDNAs, establishment of stable cell lines that contained these cDNAs. Expression of proteins was detected with immunoblots. Flow cytometric analyses were used to verify the presence of the active and inactive complexes with different antibodies. In addition, a fibrinogen-binding assay was used to determine the inhibition of the active complex by the peptide.
Results and discussion
A stable cell line of the co-transfected cDNAs of αIIb, β3 wild type and mutants of β3 (scrambled sequence of the peptide region, replacement of C499A and C512A), expressing the inactive complex on CHO cells, has allowed us to examine the important role of the peptide sequence and the cysteine residues within the peptide. The peptide inhibits the active complex formation and thereby inhibits the binding of FITC-PAC-1 in a dose dependent manner by flow cytometric analyses, as well as binding of [3H]-fibrinogen. In addition, creation of a second stable cell line containing wild type αIIb and the mutated region of β3 (residues 499–513) shows that the binding of FITC-PAC-1 and [3H]-fibrinogen on the mutant activated complex was much lower than the wild type activated complex. Our results indicate that the region 499–513 in β3 is one of the important sites for αIIb/β3 active complex formation and the cysteines play an important role in the process.
Introduction
AlphaIIbbeta3 (αIIb/β3) complex plays an important role in platelet aggregation. The αIIb/β3 complex exists in the inactive and active conformational states. The activated complex serves as ligand-binding sites (Luo et al. 2004; Schwartz et al. 1995) for four macromolecules (fibrinogen, von Willebrand factor, fibronectin and vitronectin). The binding of fibrinogen to the activated αIIb/β3 complex mediates the final step of platelet aggregation. In damaged vessel walls, αIIb/β3 undergoes a conformational change from an inactive to an active state that is able to initiate thrombi formation.
Many studies have proposed that the interaction between αIIb and β3 reside in several regions (Filizola et al. 2004; Feuston, 2003 and D'Souza et al. 1994). There are two discrete peptides defined from β3 with amino acid residues of 211–222 and 217–230, which have been reported to inhibit fibrinogen binding and affect platelet aggregation (Charo et al. 1991 and Steiner et al. 1993). In addition, two complex forming sequences of αIIb and β3 (αIIb: 94–314 and β3: 118–131, and αIIb: E117 and β3:R214), which correlate with both ligand and cation binding within the αIIb/β3 complex, have been identified (Feuston, 2003; D'Souza et al. 1994; D'Souza et al. 1991 and Charo et al. 1991).
Other investigators (Kashiwagi et al. 1999; Sun et al. 2002; Beglova et al. 2002 and Butta et al. 2003) have addressed the important function of cysteine residues in β3. The consensus is that cysteine residues play an important role in the formation of the active complex. Wang et al. (1997) have found that disruption of the disulfide bond between Cys 406 and Cys 665 of β3 did not affect αIIb/β3 ligand binding. However, in some cases, disruption of certain disulfide bonds resulted in the formation of an active complex. Kashiwagi et al. (1999) created a single amino acid mutation in the extracellular cysteine-rich repeat region of the β3 subunit (T562N), and activated αIIb/β3. Although, many laboratories have studied the extracellular, transmembrane, cytoplasmic regions and carboxyl-terminal of β3 in relation to the formation of the functional complex, the exact site remains to be determined.
Previously, we have reported that a platelet 90-kDa glycoprotein (GP 90) is involved in collagen-, epinephrine-, and thrombin-platelet interaction (Chiang et al. 1989). We have identified the protein as β3 by using two-dimensional gel (2D gel) electrophoresis, immunblotted with anti-90-kDa and anti-β3 antibodies, and Matrix Assisted Laser Desorption/Ionization-Time of Flight (MALDI-TOF). Searches of the SWISS-PROT database for the identity with the mass spectra of the trypsinized fragments, isoelectric point, and molecular weight resulted in a match with human platelet β3. Recently, we have defined a region of β3 (residues 499–513, named P4), which inhibits antagonists-induced platelet activation by inhibiting FITC-PAC-1 binding. A chemically synthesized peptide also inhibits ADP-, type I collagen- and type III collagen-induced platelet aggregation in a dose-dependent manner through the inhibition of αIIb/β3 complex formation (Chiang et al. 2005).
In the present study, we have constructed three mutant cDNAs of β3 by scrambling the amino acid residues and the other two by substituting the cysteine residues with alanine (C499A and C512A) of the P4 peptide region and expressing them with wild type αIIb, as well as expressing αIIb/β3-wild type in Chinese hamster ovary cells (CHO). We examined the binding ability of the mutant compared with wild type to FITC-PAC-1 and [3H]-fibrinogen. Results show that co-transfection with the mutated cDNAs expresses an inactive complex and binds less FITC-PAC-1 as compared to wild type. In addition, a chemically synthesized peptide (of the defined region) inhibited the binding of FITC-PAC-1 as well as inhibiting the binding of [3H]-fibrinogen onto Mn2+ -activated wild type αIIb/β3 expressed in CHO cells. These results suggest that the P4 peptide of β3 and its cysteine residues play an important role in the formation of the active complex of αIIb/β3.
Material and Methods
Reagents
Monoclonal anti-β3 was purchased from R&D systems, Inc. (Minneapolis, MN) and monoclonal anti-αIIb (clone SZ22) was purchased from Beckman Coulter (Fullerton, CA). The monoclonal antibody, which recognizes the inactive complex of αIIb/β3, AP2, was purchased from GTI Diagnostics (Waukesha, WI). The Chinese hamster ovary (CHO-K1) cell line was purchased from America Type Culture Collection (ATCC) (Manassas, VA). The transfection reagent, Lipofectamine 2000 and fluorescent dye Alexa Fluor 647 were from Invitrogen (Carlsbad, CA). All other chemicals were from Sigma Chemical Co. (St. Louis, MO), Fisher Scientific (St. Louis, MO), Bio-Rad (Hercules, CA), and Pierce (Rockford, IL). GP IIb cDNA construct is a gift from Dr. Peter Newman (The Blood Center of Southeastern Wisconsin, Inc. Milwaukee, WI).
Preparation of platelet-rich plasma (PRP)
Human blood (9 parts) from normal volunteers, were collected following an overnight fast, in polypropylene tubes containing 1 part 3.8% sodium citrate. PRP was prepared by centrifuging the citrated blood at room temperature for 10 minutes at 226 × g (Chiang et al. 1976). Whole blood and PRP were exposed to plastic surfaces or siliconized vessels only. Platelet counts of the PRP ranged from 200,000 to 300,000 per mm3.
The washed platelets for binding assays were prepared by gently mixing equal volumes of PRP and 20 mM Tris-HCl-130 mM NaCl-1 mM EDTA, pH 7.4 (Tris-EDTA), centrifuged at 1,000 × g for 5 min, washed once with Tris-EDTA and then resuspended in the Tyrode's buffer.
Binding of [3H]-fibrinogen to washed human platelets
Fibrinogen was labeled with [3H]-formaldehyde as described by others (Whitnack et al. 1985; Rice et al. 1971 and Grinnell, 1980). Briefly, human fibrinogen was dissolved in PBS at a concentration of 3.4 mg of clottable protein per ml (10 μM), dialyzed against PBS to remove salts, and stored at -20 °C. Fibrinogen was dissolved in 0.2 M sodium borate (pH 7.4) at a concentration of 10 μM and dialyzed against the same buffer. The fibrinogen was then subjected to reductive N-methylation at 4 °C with 0.1 volume of a 1% aqueous solution of [3H]-formaldehyde, followed by 0.3 volume of sodium borohydride. The labeled fibrinogen was dialyzed against PBS, assessed for amount of bound radiolabel, assayed for binding capacity and finally, stored at –20 °C.
The binding mixture consisted of washed human platelets (108) in Tyrode's buffer (5 mM HEPES, 2 mM MgCl2, 0.3 mM NaH2PO4, 3 mM KCl, 12 mM NaHCO3, 0.1% glucose, 0.1% BSA, and 1 mM CaCl2, pH 7.0). Various amounts of P4 and scrambled P4s peptides (0–80 μM) were used to determine the dose response of inhibition of binding by fibrinogen for 30 minutes at room temperature. Initiation of the assay began with the addition of ADP (final concentration of 0.5 μM), incubated at room temperature for 5 minutes, followed by adding [3H]-fibrinogen with further incubation at room temperature for an hour. Then unbound [3H]-fibrinogen was removed by washing with Tyrode's buffer three times, the pellet was then suspended in the same buffer and transferred to scintillation vials containing 7.5 ml of scintillation solution (ScintiVerse BD, Fisher Scientific, Pittsburgh, PA). Radioactivity was assessed with a Packard Liquid Scintillation Analyzer (Perkin Elmer Life and Analytical Sci., Inc., Boston, MA).
Construction and expression of wild type and mutant β3 cDNAs
Construction of a plasmid encoding for the full-length cDNA of platelet β3 were from several cDNA fragments deposited at ATCC and subcloned into the vectors pET-24a and pcDNA3.1. The β3-mut cDNA is a scrambling of the peptide sequence from 499–513 (473–487 according to numbering by Kamata et al. 2001) of β3 by PCR. The two sets of primers used were, forward primer: 5‘-ACAGATCTTGCGAGTATTCCGAGCAGTGCCCCCGGGAGGGTCAGCC-3 and reverse primer: 5‘- CAAGATCTGTCGCTAGGCTGCTCGTCCTCACACTGGGATCCCAGC-3’. The following primers were used to construct the cDNA with C499A using forward primer: 5‘-GGATCCCAGTGTGAGGCCTCAGAG-3’ and reverse primer: 5‘-CTCTGAGGCCTCACACTGGGATCC-3’ and the second cDNA with C512A using forward primer: 5‘-GCAGGACGAGGCCAGCCCCCGG-3’ and reverse primer: 5‘-CGGGGGCTGGCCTCGTCCTGC-3’. The template used for all PCR reactions was β3-wild type in pcDNA3.1. All of the final PCR products carrying the desired mutations in the full-length cDNA were treated with Dpn I, purified, transformed into DH5α, selectively screened, and sequence verified before proceeding with expression.
Peptides for inhibition studies
The first chemically synthesized peptide is C(Acm)SEEDYRP SQQDEC(Acm)S, which we referred to as P4. The second chemically synthesized peptide is the scrambled version of P4, SDC (Acm)EYPESEQRSQDC(Acm), which served as a control and is referred to as P4s.
Establishment of stable cell lines for eukaryotic expression in CHO cells and immunoblot analyses
Wild type β3 or mutant β3 (β3-mut) (4 μg of each construct) and wild type αIIb (4 μg) were co-transfected into CHO cell using Lipofectamine 2000 according to the manufacturer's specifications. Cells were incubated 37 °C for 48 hours following transfection, allowing for transgene expression in selective media containing 700 μg/ml of Geneticin for two weeks. Selected single colonies were expanded to 24-well plates and the selected integrant was screened by flow cytometry in a BD FACS Calibur using the AP2 antibody conjugated to the fluorescent dye (Alexa Fluor 647). Following selection, the cells are maintained in selective medium with 700 μg/ml of Geneticin. Immunoblot analysis with both, αIIb and β3 antibodies also verified the successful co-transfections.
Flow cytometry analysis of activated of αIIb/β3 complex
Stable cells (wild type αIIb/β3 and αIIb/β3-mut) were harvested with 0.5 mM EDTA/PBS pH 7.4, washed twice with 50 mM HEPES/1% glucose/2 mM Ca2+/2 mM Mg2+ pH 7.4 (HEPES buffer I) and resuspended in 50 mM HEPES/1% glucose/1 mM Ca2+/1 mM Mg2+ pH 7.4. (HEPES buffer II). Activation of the washed cells (1 × 106/ml) were initiated by the addition of 2 mM Mn2+ for 45 minutes (Litvinov et al. 2004), then incubated with FITC-PAC-1 for an additional 60 minutes at room temperature. Following three washes with HEPES buffer II, the cells were resuspended in the same buffer and the amount of FITC-PAC-1 binding was analyzed by flow cytometry.
Inhibition of αIIb/β3 active complex by peptides
The chemically synthesized peptides, P4 and P4s, were used to determine whether they could inhibit the activation of the wild type αIIb/β3 complex. Again, cells were prepared as previously stated, incubated with varying amounts of peptide, (either P4 or P4s, 0–80μm) and 2 mM Mn2+ for 45 minutes, followed by incubation with FITC-PAC-1 for an additional 60 minutes at room temperature. Following immunostaining, all cells were washed three times in HEPES buffer II, resuspended in the same buffer and binding was analyzed by flow cytometry.
Binding of [3H]-fibrinogen to stable co-transfected CHO cells
Binding mixture consisted of co-transfected CHO cells (either wild type or mutant, 5 × 106) in Tyrode's buffer. The assay was initiated by adding Mn2+ to final a concentration of 2 mM, incubated at room temperature for 45 minutes, then added of P4 or P4s, followed by the addition of [3H]-fibrinogen and incubated at room temperature for an additional hour. Following incubation, the bound and unbound [3H]-fibrinogen, were separated by washing with Tyrode's buffer three times, resuspended in the same buffer and transferred to scintillation vials containing 7.5 ml of scintillation solution. Radioactivity was determined with a Packard Tri-Carb 2000CA Liquid Scintillation Analyzer (Perkin Elmer Life and Analytical Sci., Inc., Boston, MA). The effect of peptides, P4 and P4s, on [3H]-fibrinogen binding followed the same binding assays as previously stated, with the exception that varying amounts of each peptide are incubated with the Mn2+.
Results
Binding of [3H]-fibrinogen on washed human platelets
We have established the optimum binding conditions [(time (one hour), room temperature, number of platelets (108) or co-transfected CHO cells (5 × 106), and concentration of fibrinogen (0.5 μM)] for the binding assay (data not shown). Using the established conditions, we tested the effects of P4 and P4s peptide on the binding of [3H]-fibrinogen on washed human platelets. The amount of [3H]-fibrinogen binding to the platelets was reduced by P4 (Fig. 1, line with diamonds). The percent inhibition by P4 is dose-dependent. Treatment with P4s did not significantly reduce fibrinogen binding (Fig. 1, line with filled squares). In addition, the P4 peptide could not inhibit the binding of fibrinogen to platelets without the addition of suboptimal concentrations of ADP (data not shown). These results suggest that the peptide probably binds on GP IIb to inhibit the αIIb/β3 active complex formation rather than directly affecting the fibrinogen binding to the activated complex.
Inhibition of [3H]-Fibrinogen binding to human platelets by P4 and scrambled P4 (P4s) peptides. Binding assay of [3H]-fibrinogen to human platelets in the presence of various amounts of P4 (line with filled diamonds) and scrambled P4 peptides (line with closed squares) 0–80 μM for 30 minutes at room temperature and assessed for radioactivity. The data expressed is an average of duplicates per experiment and repeated three times with similar results. A representative study is shown.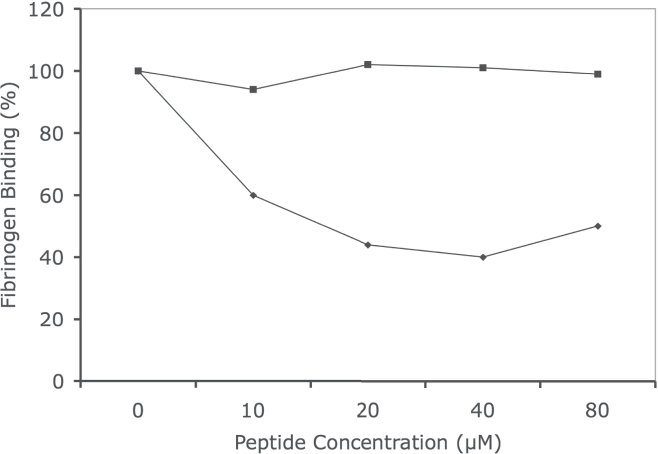
In order to study the effects of P4 (wild type β3) on the formation of active αIIb/β3, we have engineered a construct containing the scrambled sequence of the active peptide (P4s) in β3 cDNA. It served as a control (named as β-mut) in co-transfection with wild type αIIb cDNA into CHO cells and proved that the P4 peptide is an inhibitor of αIIb/β3 active complex formation.
Recombinant proteins of αIIb, β3, and β3-mut expressed in CHO cells
Expressions of co-transfected proteins, αIIb with wild type-β3 or β3-mut were examined by immunoblot analyses. Figure 2 shows that both wild type αIIb and wild type β3 proteins and wild type αIIb and β3-mut were detected in co-transfected CHO cell lysates. Panels A and B shows a set of immunoblots of co-transfected CHO cells (lane 1: mock, lane 2: αIIb/β3 wild type, and lane 3: αIIb/β3-mut). The αIIb antibody recognized αIIb expressed in CHO cells expressing wild type αIIb/β3 (lane 2) and αIIb/β3-mut (lane 3) as shown in panel A (arrow). In addition, the β3 antibody recognized β3 proteins expressed in the wild type αIIb/β3 (lane 2) and less in αIIb/β3-mut (lane 3) as shown in panel B (arrow).
Immunoblots of wild type αIIb, β3 and β3-mut proteins. The wild type cDNAs of αIIb and β3 as well as αIIb/β3-mut were co-transfected into CHO cells allowing for transgene expression 48 hours following transfection. The cells are harvested, washed, lysed, and protein concentration determined. Equal amounts of protein were separated by SDS-PAGE and electroblotted for immunoblot analysis. Panel A and B are 10% and 7.5% reduced SDS-PAGE, respectively. Monoclonal antibodies (anti-αIIb-panel A and anti β3-panel B, preabsorbed with CHO cells) are used to detect the presence of each integrin. Lane 1, Mock control; Lane 2, Co-transfected cells of wild type αIIb/β3 cDNAs; Lane 3, Co-transfected cells of wild αIIb and β3-mut cDNAs. Arrows indicates αIIb protein in panel A and β3 protein in panel B.
We have then used flow cytometry to study the αIIb/β3 expression in co-transfected CHO cells. Figure 3, panel A showed the presence of an inactive complex, identified by the AP2 antibody in both αIIb/β3 (line 2) and αIIb/β3-mut (line 3). The binding capacity of AP2 (calculated as percentage gated subset) was similar between wild type β3 and β3-mut. The mutation made in β3 did not interrupt the ability for the protein to be expressed, as well as being transported to the cell surface and forming the complex. Panel B showed that in the absence of Mn2+, the FITC-PAC-1 binding level (calculated as percentage gated subset) was also similar in both wild type (line 2) and αIIb/β3-mut expressed complexes (line 3). However, in panel C, following incubation with 2 mM Mn2+, the ability to bind FITC-PAC-1 increased in the αIIb/β3 wild type expressed complex (line 2) but not the αIIb/β3-mut expressed complex (line 3).
Analysis of expressed αIIb/β3 wild type and αIIb/β3-mut by flow cytometry. Stable lines expressing wild type or mutant β3 and wild type αIIb were incubated with different monoclonal reagents. 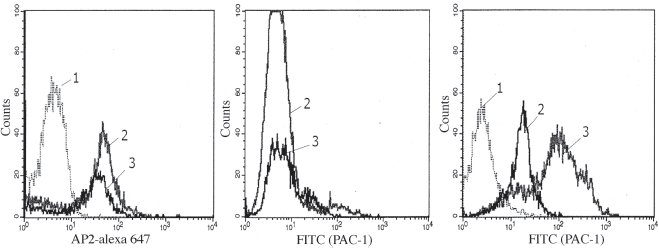
In addition to the wild type β3 and β3-mut, we have constructed two other mutants to examine the role of the cysteine residues within the peptide. We have performed replacement of cystine residues (499 and 512) one at a time with alanine. Table 1 shows following Mn2+ treatment, the wild type αIIb/β3 had a higher percentage (92%) of PAC-1 binding, compared to a lower percentage with the αIIb/β3-mut (61%). Replacement of either 499 (89%) or 512 (91%) with Ala, the binding of PAC-1 did not change significantly compared to wild type. The binding of PAC-1 by GP IIb/IIIa-C499A (89%) or GP IIb/IIIa-C512A (91%) did not change significantly compared to wild type. However, these results do indicate that Mn2+ could not fully activate αIIb/β3-mutants resulting in fewer numbers of the formation of the active complex and thus less binding of FITC-PAC-1.
We have assayed and analyzed the effect of both the P4 and P4s peptides to the Mn2+-activated αIIb/β3 complex by measuring the binding of FITC-PAC-1. Figure 4 shows the effects of the addition of the P4 peptide. Panel A shows the control CHO cells while panel B represents the FITC-PAC-1 bound to Mn2+-activated αIIb/β3 complex. When cells are pre-incubated with P4 and Mn2+, the binding of FITC-PAC-1 is inhibited in a dose-dependent manner. The percentage of inhibition is 28% (panel C, 60 μM) and 60% (panel D, 80 μM), respectively. The concentration of peptide required for inhibition was similar to that used in washed human platelets (Schwartz et al. 1995). The P4s did not show any significant inhibition on the binding to the Mn2+-activated αIIb/β3 complex (data not shown).
Effect of the defined β3 peptide (P4) on the binding of FITC-PAC-1 to co-transfected CHO cells. The CHO cells expressing αIIb/β3 inactive complex were activated with 2 mM Mn2+ in the presence of various amounts of P4 for 45 minutes at room temperature. Following incubation, an aliquot of PAC-1-FITC was added and incubated at room temperature for an additional 60 minutes. Following three washings, the fluorescent stringency was detected by flow cytometry. 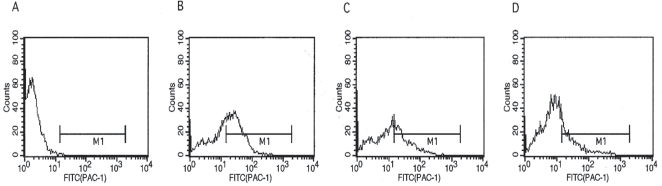
We have established the optimal conditions for [3H]-fibrinogen binding assays [fibrinogen final concentration (80 nM, cell number (5 × 106), temperature, and incubation time (60 minutes)] to determine the effects of the P4 and P4s peptides. We investigated the ability of the P4 peptide to inhibit fibrinogen binding to αIIb/β3 in the presence of 2 mM Mn2+. The P4s peptide was used as the control in the [3H]-fibrinogen binding assays. The addition of P4 peptide decreased the amount of fibrinogen binding to the Mn2+-activated αIIb/β3 complex in a dose-dependent fashion (Fig. 5, line with open circles), as the peptide dose increased, the amount of fibrinogen binding to the cell surface decreased. The scrambled peptide, P4s, did not inhibit the fibrinogen binding significantly (Fig. 5, line with filled squares), indicating that the order of amino acid residues in the P4 peptide sequence is important for inhibiting αIIb/β3 complex formation, thus inhibiting fibrinogen binding. Although, this experiment may not represent the cellular physiology and biochemistry of human platelets, it may be another useful tool to study the interaction processes.
Binding of [3H]-fibrinogen on CHO cells expressing the αIIb/β3 complex and the effect of the P4 peptide (open-circle line) and the P4s (square-filled line) from β3. The cells were incubated with 2 mM Mn2+ and different amounts of peptide (0–80 μM). The Y-axis indicates the percentage of [3H]-fibrinogen binding. The peptide concentrations were 10 μM, 20 μM, 40 μM and 80 μM for various time points beginning with time 0, respectively (P4, line with open circles; P4s, line with filled squares). The data expressed is an average of duplicates per experiment and repeated three times with similar results. A representative study is shown.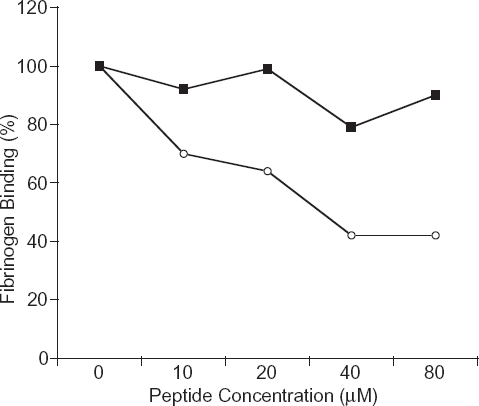
Discussion
We have previously defined a peptide (P4), amino acid residues from 499–513 of β3 as one of several important sites of the active conformational state of the complex αIIb/β3 (Chiang and Zhu, 2005). In this investigation, we have performed experiments to study its function on the formation of the αIIb/β3 active complex. We have established a stable cell line that was co-transfected with wild type αIIb and wild type β3 cDNAs in CHO cells. Results revealed that the P4 peptide inhibited both FITC-PAC-1 and [3H]-fibrinogen binding to the cell surface expressed Mn2+-activated αIIb/β3 complex. In addition, three stable cell lines of co-transfected wild type αIIb and β3-mut cDNAs also demonstrated a decrease in FITC-PAC-1 binding and [3H]-fibrinogen binding. These results are consistence with our earlier report that the P4 peptide inhibits binding of FITC-PAC-1 on human platelets (Chiang and Zhu, 2005).
Collagen-induced platelet aggregation is mediated by the released of ADP from the collagen-activated platelets. In the present studies, we have used suboptimal concentrations of ADP to trigger partial platelet activation for the binding of [3H]-fibrinogen. However, under these same conditions, the addition of P4s does not inhibit the binding of [3H]-fibrinogen. In contrast, the addition of P4 after platelets become fully activated, does not inhibit fibrinogen binding. The 2,3,5,6-tetrafluorophenyl ester (purchased from Molecular Probes)-labeled P4 and P4s could not bind on the fibrinogen-coated wells. These findings suggest that the peptide does not bind to fibrinogen per se to lower [3H]-fibrinogen concentration in turn to decrease the binding.
Parise et al. (1987) and Du et al. (1991) have reported that the RGD peptide (a recognition sequence for fibrinogen binding) or RGD-derived peptides inhibit(s) the binding site exposed by conformational changes in platelets. Our results showed that P4 peptide inhibits the binding of both FITC-PAC-1 and [3H]-fibrinogen on the stably co-transfected Mn2+-activated αIIb/β3 complex. The dose-dependent inhibitory effect of P4 peptide indirectly leads us to postulate that the peptide binds to αIIb and prevents the formation of the active αIIb/β3 complex. Alternatively, P4 modifies the αIIb/β3 complex conformation in some fashion to keep it in the inactive state. In our results, the peptide inhibitory effects are partial. A plausible explanation would be that there is more than one region involved in the activation of the complex formation. Our results would support Takagi's finding, in which he suggests a “2-site docking model” from his study of ligand recognition by RGD-dependent integrins (Takagi, 2004).
Blystone et al. (1995) has reported that exposure to Mn2+ shifts the β3 integrin to their high affinity state and possibly activation of the receptor or the subsequent function of β3 following activation. In addition, Takagi et al. (2002) have reported that integrins, αVβ3 and αIIbβ3, have a highly bent conformation and had low affinity for biological ligands under physiological condition. The addition of Mn2+ resulted in a switchblade-like opening to an extended structure that has high affinity for biological ligands. Our results are consistence with theirs in that upon exposure to Mn2+, the αIIb/β3 complex shifts from its inactive to the active state as shown by the increase in FITC-PAC-1 binding.
Cysteine residues in β3 play important roles in αIIb/β3 complex activation. Sun et al. (2002) reported that disruption of the long-range β3 Cys5-Cys435 disulfide bond resulted in the production of constitutively active αIIb/β3 integrin complex. Another report proposed that the cysteine residues located from 616–690 of the carboxyl-terminal region was important in enhancing ligand binding (Butta et al. 2003). Kashiwagi et al. (1999) found that disrupting only a single disulfide bond in the cysteine-rich repeat region of β3 was enough to activate αIIb/β3. The P4 peptide, which we have identified is present within the cysteine-rich region of β3 and contains two cysteine residues, which may form a disulfide bond with other cysteines of αIIb. We used the DIpro 2.0 software to predict the location, which the disulfide bond formation would reoccur (Cheng et al. 2006). According to the software results of proposed β3-mut, the predicted disulfide bridge reforms at 513–527. Altering the positions of these two cysteine residues may affect the ability of Mn2+ to activate αIIb/β3. The β3-mut cDNA we created and expressed in CHO cells was not clearly defined by Coomassie Brilliant blue stained SDS-PAGE or by immunoblot analysis with an anti-β3 monoclonal antibody (Fig. 1, panel B, lane3). However, with flow cytometry, we observed the presence of the inactive complex of αIIb/β3-mut with AP2 (an antibody, which recognizes the αIIb/β3 inactive complex). The percentage-gated subset of AP2 binding was similar between the two different stable lines αIIb/β3 and αIIb/β3-mut. These results indicate that expression and transport of the αIIb/β3-mut proteins to the cell surface does occur. The poor immunoblot result of β3-mut may be a consequence of the inability of the antibody to recognize the mutated sequence of amino acid residues. The αIIbβ3-mut cDNA expressed in CHO cells did not show strong activation of the inactive complex with the addition of Mn2+. Our results of substitution of C499A and C512A resulted in less PAC-1 binding suggesting these two cysteines are also important.
Integrins have become attractive therapeutic targets. Many drugs, which inhibit integrin αIIb/β3 binding can effectively block agonists induced platelet aggregation in vitro. However, intravenous infusion or oral ingestion of these drugs failed to effectively block pathological thrombosis in certain patients. This raises a question of “How effective is the ligand-mimetic integrin blockade?” Our results demonstrates that the P4 peptide is important for interfering with αIIb/β3 activation, thus inhibiting binding of fibrinogen on activated platelets and platelet aggregation in vitro, may be another candidate as a therapeutic agent. However, its functional significance in vivo is not clear and requires further investigation.
Abbreviations
platelet-rich plasma
20 mM Tris-HCl-130 mM NaCl-1 mM EDTA, pH 7.4
5 mM HEPES, 2 mM MgCl2, 0.3 mM NaH2PO4, 3 mM KCl, 12 mM NaHCO3, 0.1% glucose, 0.1% BSA, and 1 mM CaCl2, pH 7.0
50 mM HEPES/1% glucose/2 mM Ca2+/2 mM Mg2+ pH 7.4
Fluroscence isothiocyanate conjugated anti-GP IIb/IIIa
Chinese hamster ovary cell.
Footnotes
Acknowledgements
The authors wish to thank Mrs. X. - R. Fang for her expert technical assistance, Dr Zhu for discussion, and Dr. Peter Newman, The Blood Center of Southeastern Wisconsin, Inc. Milwaukee, WI for a αIIb construct. The Office of Biomedical Laboratory Research, Department of Veterans Affairs supported the present investigation.