
Abstract
Multiple sclerosis (MS) is the most common demyelinating disease of the central nervous system (CNS). Without myelin, nerve impulses in the CNS are slowed or stopped, leading to a constellation of neurological symptoms. Demyelination also provides a permitting condition for irreversible axonal damage. Remyelination of MS lesions largely fails, although oligodendrocyte precursors and premyelinating oligodendrocytes (myelin forming cells) are present in many demyelinated plaques. Insulin-like growth factor (IGF)-1 is a growth factor that should provide the appropriate signals to promote repair of MS lesions, because it acts as a survival factor for cells of the oligodendrocyte lineage and stimulates myelin synthesis. In a pilot study on MS patients, no detectable remyelinating effects in the CNS were observed following subcutaneous administration of IGF-1. A number of reasons might explain a lack of beneficial effects: a) it is unlikely that subcutaneous administration of IGF-1 provides sufficient passage across the blood-brain-barrier and into the CNS, b) the biological actions of IGF-1 are tightly regulated by several insulin-like growth factor binding proteins (IGFBPs), which become upregulated in the demyelinated lesions and may prevent access of IGF-1 to its receptor, c) IGF-1 not only acts on oligodendrocytes, but also stimulates the proliferation of astrocytes, which form the glial scar that impedes repair processes. In this review, we will discuss strategies to enhance IGF-1 signaling in the CNS utilizing a) alternative routes of administration, b) IGF analogues that displace IGF-1 from regulatory IGFBPs and c) strategies to selectively target IGF-1 to oligodendrocytes.
Keywords
Introduction
Multiple sclerosis (MS) is a chronic multifocal demyelinating disease of the central nervous system (CNS) of unknown etiology. One in thousand persons in Europe are afflicted with this disease which generally occurs between the ages of 30–40 (Noseworthy et al. 2000). Pathological hallmarks of MS are inflammation, demyelination, degeneration and loss of oligodendrocytes, proliferation of astrocytes, and axonal damage in the CNS (Wolswijk, 2000). Figure 1 shows a chronic MS lesion, characterized by demyelination and astrogliosis. The underlying cause of myelin destruction and death of oligodendrocytes in MS is not completely understood, however data suggest that MS may be a disease of autoimmune nature (Steinman, 1996). Disruption of the myelin sheaths, axonal injury and glial scar formation is responsible for the clinical symptoms that accumulate in the course of disease. For a disease with such a high impact, the present therapeutic options are disappointing with restricted clinical benefits.
Luxol-fast blue staining (myelin staining) of the white matter from MS (A). The pale area is a demyelinated plaque (black arrows) and the surrounding non-affected area is myelinated white matter (white arrows). GFAP-staining of astrocytes in a MS plaque (B) showing the presence of large reactive astrocytes in MS (black arrows) and normal astrocytes surrounding the MS plaque (white arrows). Astrocytes in the MS plaques are stained using an antibody against glial fibrillary acidic protein (GFAP) a specific marker for astrocytes.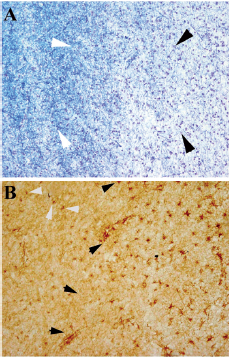
In order to prevent further damage from occurring in the CNS, current therapies target the immune system and are aimed at reducing relapses, which are caused by the formation of new demyelinating lesions. Current immunomodulatory therapies, including interferon-γ (Fillipini et al. 2003) and glatiramer acetate, show only a modest protective effect against relapses. Therapies directed at repairing damage, e.g. based on remyelination strategies, have not yet been developed.
Following demyelination, some remyelination of MS lesions can be observed during the early stages of the disease (Prineas et al. 1993), although this is often limited in its extent, and largely fails as the disease progresses. Most chronic lesions of MS demonstrate no remyelination, although oligodendrocyte precursor cells and premyelinating oligodendrocytes are present in many demyelinated plaques (Chang et al. 2002; Scolding et al. 1998; Wolswijk, 2002). This suggests that the microenvironment in chronic MS lesions may possess the potential for remyelination, yet failure of this event is likely due to a lack of appropriate signals to stimulate remyelination. Reduced (neuro) trophic support to oligodendrocytes might be implicated in oligodendrocyte apoptosis and the failure of remyelination in MS. Insulin-like growth factor (IGF)-1 is a (neuro)trophic growth factor with insulin-like metabolic activities which possesses potential clinical applications, particularly in (neuro) degenerative disorders of the CNS.
Short Overview of the IGF System
The components of the IGF system include IGF-1 and -2 (IGFs), type-1 and -2 IGF receptors and six insulin-like growth factor binding proteins (IGFBPs). In the CNS, both IGF-1 and -2 are produced as paracrine and autocrine hormones. IGF-2 is genetically related to IGF-1 and both hormones display approximately 62 percent sequence homology. Whereas IGF-1 expression in the CNS is high in neuronal rich regions of the brain (Bondy et al. 1992), IGF-2 is highly expressed in mesenchymal support structures of the CNS, including the choroid plexus (Logan et al. 1994).
Biological actions of these growth factors on target cells are mediated by cell-surface receptors. Two types of IGF receptors have been identified in human brain, the type-1 and type-2 IGF receptor. The type-1 IGF receptor is a membrane glycoprotein consisting of two α-subunits and two β-subunits (Yamasaki et al. 1993). The α-subunit is entirely extracellular and contains the ligand-binding site. The β-subunit contains a transmembrane domain with a short extracellular region, and a tyrosine kinase domain in its cytoplasmic portion. The type-2 IGF receptor is structurally and functionally quite different from the type-1 IGF receptor. The type-2 IGF receptor consists of a single glycosylated polypeptide. The type-2 IGF receptor lies primarily extracellular with a short cytoplasmic tail and consists of 15 repeated mannose-6-phosphate (M6P)-binding units (Kornfeld et al. 1992). The major functions of the type-2 IGF receptor binding appear to be lysosomal enzyme trafficking (Kornfeld et al. 1992) and IGF-2 degradation via receptor mediated-internalization (Morgan et al. 1987). Type-2 IGF receptors are not thought to be involved in cell signaling. There is ample consensus today that the biological actions of both IGF-1 and -2 are mediated through type-1 IGF receptors.
In vitro and in vivo, IGF-1 stimulates DNA synthesis and cell growth. A cellular action of IGF-1 that is complementary to its stimulation of cell proliferation is its capacity in certain cells to inhibit apoptosis. IGF-1 also induces differentiation of neurons and oligodendrocytes (Feldman et al. 1997). Two major pathways induce the actions of IGF-1 and IGF-2 through type-1 IGF receptors, the first is the mitogen-activated protein (MAP) kinase pathway and the second is the serine-threonine protein kinase Akt pathway. The MAP kinase pathway is often associated in proliferation and differentiation (Kim et al. 1997), whereas the Akt pathway is induced in IGF-1 mediated cell survival (Brunet et al. 1999) and protection from apoptosis (Kermer et al. 2000).
Biological actions of IGF-1 and IGF-2 are modulated through six IGFBPs. These proteins contain high affinity binding sites for IGF-1 and IGF-2, and they possess an 80% sequence homology with each other (Rajaram et al. 1997). In the circulation, the major IGFBP form is IGFBP-3, which binds IGF-1 and IGF-2 with similar affinities (Baxter, 1994). Circulating IGFBP-3 inhibits insulin-like activity, regulates the rate of transport and prolongs half-lives of plasma IGF-1 and IGF-2. As carrier proteins of IGF-1 and IGF-2, a major function of IGFBPs is to transport and target IGFs to specific tissues and cell types. Therefore, these proteins have a central position in IGF ligand-receptor interactions by influencing bioavailability and extracellular distribution. IGFBPs are able to enhance or inhibit IGF effects by regulating binding to type-1 IGF receptors, a mechanism that is thought to be dependent on specific cell and tissue properties (Firth and Baxter, 2002). Binding of IGF/IGFBP complexes to components of the extracellular matrix (ECM) and the cell-surface can facilitate the release of IGFs leading to enhanced delivery to type-1 and type-2 IGF receptors. For example, the binding of IGFBP-2 to chondroitin-6-sulphate, an ECM component, decreases the binding affinity of IGFBP-2 to IGF-1 by 3-fold in rat brain, leading to increased levels of biologically active IGF-1 (Russo et al. 1997). Another mechanism involved in IGF regulation are specific proteases, which are secreted by cells and act as growth stimulators by increasing local IGF availability (Conover et al. 1993). Circulating and tissue specific IGFBPs bind IGF-1 and IGF-2 by forming biologically inactive IGF/IGFBP complexes. IGF-1 and -2 can be released from these complexes, for example by proteolysis of IGFBPs. IGFBP fragments generated by the action of cellular proteases show a marked loss of IGF binding affinity.
In conclusion, IGFBPs can influence IGF actions in different ways. The multitude of these effects depends on the cell type in which they are expressed, how they interact with cell surfaces and ECM, the presence of specific proteases and the formation of high affinity binding proteins complexed with IGFs.
The Role of Insulin-Like Growth Factor-1 in Multiple Sclerosis
During nervous system development, IGF-1 plays a crucial role in cell proliferation, differentiation, and survival (Bondy and Cheng, 2004; Russo et al. 2005). During that period IGF-1 and type-1 IGF receptors are highly expressed in neuronal rich regions, such as the spinal cord, midbrain, cerebral cortex, hippocampus, and olfactory bulb (Anlar et al. 1999; Beck et al. 1988). We have shown that cerebral white matter in human neonates, undergoing active myelination, contains a 3-fold higher density of type-1 IGF receptors than in adults, indicating that IGF-1 also plays a important role in the myelination of the human CNS (De Keyser et al. 1994b).
In vitro experiments have demonstrated that IGF-1 greatly enhances oligodendrocyte survival (McMorris et al. 1986), myelin production (Roth et al. 1995) and proliferation of oligodendrocyte precursors (Mozell and McMorris, 1991). The effects of IGF-1 are not only restricted to oligodendrocytes of the CNS, but also apply to Schwann cells of the pheripheral nervous system, which demonstrate enhanced differentiation, myelin production, and increased survival in response to IGF-1 stimulation (Cheng et al. 1999; Ogata et al. 2004; Syroid et al. 1999).
The importance of IGF-1 in myelin production has also been demonstrated in several animal models. In the cuprizone model, demyelination occurs in the corpus callossum and superior cerebellar peduncles, and remyelination in these areas ensues when treatment with cuprizone is terminated. Cuprizone-induced demyelination in mice deficient of type-1 IGF receptors has demonstrated inadequate remyelination and lack of oligodendrocyte progenitor cells (OPC) accummulation in the site of injury (Mason et al. 2003). Transgenic mice that overexpress IGF-1 show increased brain growth and myelination (Carson et al. 1993). Myelin content in these animals was increased by 130%. By comparison, IGF-1 knock-out mice displayed reduced brain size, hypomyelination, reduced density of oligodendrocytes, loss of neuron populations, as well as reduced glucose uptake (Beck et al. 1995; Cheng et al. 2000; Liu et al. 1993). Transgenic mice overexpressing IGFBP-1, an inhibitory binding protein for IGF-1, showed reduced myelinated axons and thickness of the myelin sheets, this feature was induced by reduction in myelin expression (Ye et al. 1995). Other studies have shown that systemic application of IGF-1 in acute demyelinating experimental autoimmune encephalomyelitis (EAE), an animal model for MS, significantly reduced the number and area of the demyelinated lesions in the spinal cord (Liu and Yao, 1995; Yao et al. 1995). IGF-1 increased the number of remyelinated axons, reduced the permeability of the blood-spinal cord barrier, and enhanced myelin expression.
However, in chronic-relapsing EAE, systemic administration of IGF-1 together with IGFBP-3 after disease onset resulted in increased severity and or relapses (Lovett-Racke et al. 1998). In line with these results, long-term application of IGF-1 in chronic- relapsing EAE from the standpoint of myelin gene expression and repair, showed no positive effects of IGF-1 (Canella et al. 2000). In a clinical trial with systemically administered recombinant IGF-1 in MS patients, no effects on either new lesion formation or remyelination of existing lesions could be demonstrated (Frank et al. 2002). Data from this clinical trial, as well as from the above mentioned study on chronic relapsing EAE raise doubts about the effectiveness of systemic administered IGF-1 in chronic forms of demyelination. In the following sections, we dicuss several obstacles that might explain a lack of beneficial effects of systemic administered IGF-1.
Enhancing the Level of Free IGF-1 and Targeting IGF-1 into the CNS
One possible obstacle in an IGF-1 based therapy is the limited penetration across the blood brain barrier (BBB) that prevents free passage of large proteins into the CNS. However, there is some evidence that IGF-1 from the circulation might be transported across these barriers, albeit to a limited extent. One possible mechanism to transport IGF-1 from the circulation into the CNS is through-receptor mediated transcytosis (Reinhardt and Bondy, 1994). However, it is still not clear which receptor is involved in such transport system. It has been suggested that the presence of type-1 IGF receptors on the endothelium might play a role in such transport. The transport of peripheral IGF-1 into the CNS may also be influenced by IGFBPs which become saturable at the BBB (Weihong and Kastin, 2000). Other studies have shown that the uptake of circulating IGFs into cerebrospinal fluid (CSF) and probably into the CNS appears to be independent of type-1 IGF receptors as well as IGF-binding proteins. There is evidence that choroid plexus megalin is involved in neuroprotection by serum IGF-1 in Alzheimer disease by its dual effects on transporting IGF-1 in across the BBB, and by enhancing the clearance of brain amyloid-β (Carro et al. 2005). However, it remains questionable whether systemic levels of IGF-1 would indeed raise ligand levels in the deep white matter of the CNS in which demyelination occurs. The amount of IGF-1 eventually reaching the deep white matter will depend on the concentration of circulating IGF-1 and how avidly the BBB transport system uptakes it.
Approaches that bypass the BBB and show that IGF-1 has neuroprotective effects in the CNS of animals use surgically invasive procedures, such as the intracerebrovascular or intraparenchymal administration. These methods are not applicable to MS patients but do demonstrate a need for targeting. Intranasal administration of IGF-1 offers an alternative strategy to bypass the BBB and is a method that provides several advantages; it is a non-invasive way of drug application, there is a direct delivery of the drug into the CNS, levels of the drugs are more concentrated by avoiding the diluting effects of the circulation, and degradation and destruction of drugs are minimized by avoiding the gastrointestinal tracts. Thorne and co-workers have demonstrated that intranasal application of IGF-1 results in a rapid delivery of IGF-1 into multiple areas of the CNS, including the deep white matter in which demyelination can occur (Thorne et al. 2004). In this study, IGF-1 was delivered from the nasal cavity along olfactory and trigeminal pathways and was accompanied by activation of signaling pathways in several areas that express high levels of type-1 IGF receptors. Thorne and coworkers suggested an extracellular route of transportation from the nasal passages into the CNS associated with components of the trigeminal nerve. This mode of transport provides evidence for a rapid and direct pathway for protein transport into the CNS following intranasal administration.
A second obstacle in an IGF-1 based therapy lies in the complex regulation of IGF-actions. The biological effects of IGF-1 on target cells are mediated by interactions with the type-1 IGF receptor (LeRoith et al. 1995). However, six IGFBPs govern this interaction (Clemmons, 1997; Spagnoli and Rosenfeld, 1997). All six IGFBPs may inhibit IGF-1 actions by sequestering IGF-1, thereby preventing the interaction of IGF-1 to its receptor. We have found that IGFBP-1 and IGFBP-6 were upregulated on oligodendrocytes in the periplaque white matter in MS lesions (unpublished results). Furthermore, it has been demonstrated that these IGFBPs bind IGF-1 with high affinity, and inhibit IGF-1 induced survival and myelin production of primary oligodendrocytes (Kühl et al. 2002, 2003). These results suggest that the up-regulation of IGFBPs lead to a shortage of biologically active (free) IGF-1 in the demyelinated MS plaques, contributing to the lack of (re)-myelination. However, the exact function of IGFBP-1 and IGFBP-6 in physiological and pathological situations is yet unknown.
IGF-1 analogues that display low affinity for IGFBPs, and IGF-1 analogues that display high affinity for IGFBPs, which are able to displace endogenous IGF-1 and or IGF-2 from IGFBPs, may be suitable candidates for stimulating (re) myelination in MS (Fig. 2). Des(1–3) IGF-1 is a truncated form of IGF-1 lacking the tripeptide glycine-proline-glutamate (GPE) at the amino-terminus of the full-length form (Sara et al. 1986). The biological potency of this analogue is significantly higher than that of the full-length form and is explained by its reduced affinity for IGFBPs. IGF-1 analogues that display high affinity for IGFBPs, which can therefore displace endogenous IGF-1 from IGFBPs might also be considered. This approach has already been studied in rats with focal cerebral ischemia. In this model, the intracerebroventricular administration of the IGF-1 analogue [(Leu24,59,60, Ala
31
) hIGF-1] with high affinity to IGFBPs and no affinity to type-1 IGF receptors, increased levels of biologically active IGF-1 and provided potent neuroprotection (Loddick et al. 1998).
A schematic overview: Astrocytes regulate their own cellular action through IGF-1, and Astrocytic IGF-1 has no effect on (pre)myelinating oligodendrocytes in MS. Astrocytes become astrogliotic forming the tissue scar in MS lesions and preventing remyelination by oligodendrocytes (A). Through intranasal application of IGF-1 analogues and or IGFBP ligand inhibitors, IGF-1 is released from IGFBPs which are present on oligodendrocytes and IGF-1 become available to oligodendrocytes, resulting in remyelination (B).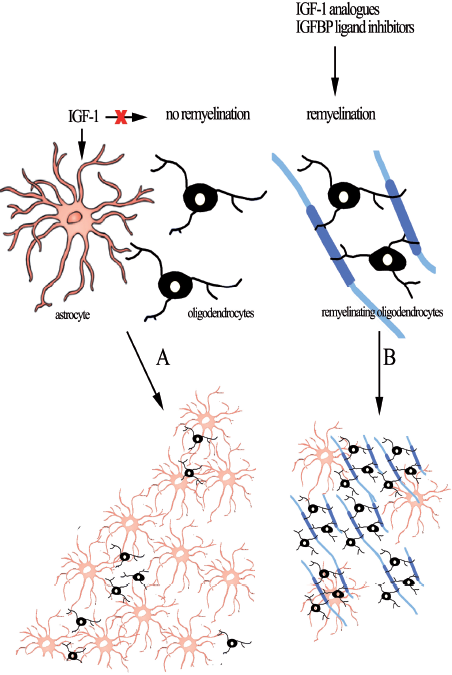
A third obstacle lies in the targeting of IGF-1 signaling to the myelin producing cells: the oligodendrocyte. Such a targeting is complicated by the high expression of type-1 IGF receptors on other cells in the CNS. We have demonstrated that type-1 IGF receptors were present on neurons and other glial cells, including microglia and astrocytes (De Keyser et al. 1994a; Wilczak and De Keyser, 1997). This may have important implications for the clinical use of IGF-1 in MS. Chronic plaques of MS contain a dense network of astrocytes, which are responsible for the characteristic astrogliotic plaque. In vitro studies have shown that IGF-1 enhances the proliferation of astrocytes (Chesik et al. 2004; Tranque et al. 1992). As acute MS lesions are rapidly invaded by reactive astrocytes, enhancing the levels of IGF-1 in MS lesions may not only protect oligodendrocytes and stimulate remyelination but also enhance the astrogliosis that creates a glial scar, limiting remyelinating processes. For these reasons, targeting IGF-1 effects to oligodendrocytes is crucial. A feasible approach for targeting oligodendrocytes consists of intervening in IGF-1 regulation by IGFBPs. In MS lesions, we have shown a unique distribution of IGFBPs expression on oligodendrocytes and astrocytes. Oligodendrocytes in demyelinated MS plaques display an upregulation of IGFBP-1 and IGFBP-6, whereas astrocytes show increased expression of IGFBP-2 and IGFBP-4 (Chesik et al. 2006). An interesting prospect is the use of non-peptide small molecules that act as specific IGFBP ligand inhibitors and prevent binding of IGF-1 to specific IGFBPs (Chen et al. 2001; Zhu et al. 2003). This approach would result in an elevation of biological active IGF-1 that is available in the vicinity of olgodendrocytes. NBI-31772 is a recently developed non-peptide small molecule that binds to all six known IGFBPs and displaces biologically active IGF-1 from all six binding proteins (Liu et al. 2001). The neuroprotective effects of NBI-31772 has been studied in experimental models of cerebral ischemia. This study has demonstrated that intracerebroventricular administration of NBI-31772 significantly reduced ischemic brain damage and infarct size. (Mackay et al. 2003). New potent ligand-inhibitors could be designed which selectively bind individual IGFBPs in order to enhance and target bioactive IGF-1.
Another approach to elevate IGF-1 levels as well as target IGF-1 into oligodendrocytes is altering the expression level of IGFBPs on oligodendrocytes and astrocytes in MS. IGFBP-2 is of particular interest because hypertrophic astrocytes in MS lesions express high levels of IGFBP-2 (Chesik et al. 2006). Astrocytes are the primary source of IGF-1 in damaged CNS, and it has been suggested that this growth factor assists in neuronal protection as well as in facilitation of myelin production. We have shown that reactive astrocytes in vitro and in situ upregulate IGFBP-2 and that combined treatment of IGFBP-2 and IGF-1 does not inhibit IGF-1 stimulated astrocyte proliferation, whereas it inhibits IGF-1 stimulated survival of oligodendrocytes (Chesik et al. 2004). We propose that an upregulation of IGFBP-2 in MS facilitates the process of astrogliosis by targeting IGF-1 to these cells. Inhibition of endogenous expression of IGFBP-2 in astrocytes by means of microRNAs (miRNAs) might have implications on cell proliferation and maturation of these cells. In MS, astrocytes become reactive and form the glial scar, which is thought to impede remyelination processes. MiRNAs are a growing family of single-stranded forms of RNA, which regulate the expression and production of proteins. Such an approach was already investigated in neuroblastoma (NB) cells (Tanno et al. 2005). Neuroblastoma is a malignant childhood tumor, in which IGFBP-5 is frequently expressed. By suppressing the expression level of IGFBP-5 in NB cells through miRNAs, NB cells become more prone to apoptosis (Tanno et al. 2005). The use of these molecules as therapeutics to influence the expression of IGFBPs during demyelination has a long way to go. However, regulating the expressing of IGFBPs through miRNAs opens a new window to selectively study the role of IGFBPs in several diseases such as MS.
Conclusion
The use of IGF-1 analogues, IGFBP ligand inhibitors and microRNAs to modify actions of IGF-1 signaling and induce remyelination in the CNS would be a tremendous breakthrough in the treatment of human demyelinating diseases, such as MS. The unique distribution of IGFBPs in MS lesions as well as a unique functional characteristic of the IGF-1 receptor on oligodendrocytes offers a means to specifically target this cell type.
Footnotes
Acknowledgments
This work was supported by the School of Behavior and Cognitative Neurosciences (BCN) Groningen and “MSanders”, The Netherlands.