
Abstract
Recent drug approvals in the field of prostate cancer therapy have brought about a change in the treatment landscape of locally advanced and metastatic castration-resistant prostate cancer. While this improvement offers a welcoming change in the standard treatment practice of prostate cancer, questions remain with regard to the proper sequencing of the right therapy for the appropriate patient. This review highlights the pre-clinical, safety and clinical studies that help bring to the forefront the drugs recently approved for the treatment of advanced prostate cancer and offer some insights to its use.
Introduction
Prostate cancer has become the most common non-cutaneous cancer in the United States (US) since the late 1980's and early 1990's generally attributed to the prostate-specific antigen (PSA) screening that detected more early-stage prostate cancers. 1 In 2011 alone, about 240,890 men will be diagnosed and about 33,720 will die of prostate cancer making it the second leading cause of cancer death in American men, behind lung cancer. 2 While most patients are cured especially when the disease is still localized and treated by radical prostatectomy and/or radiation therapy, with still about one-fifth of patients undergoing active surveillance for indolent disease, around 20% to 40% of patients who undergo primary therapy experience biochemical relapse ([PSA > 0.2 ng/mL for those who undergo surgery), and 30%–70% of those with biochemical recurrence develop metastatic disease within 10 years after local therapy.3–5
Since prostate cancer is believed to be an androgen-driven disease, initial therapy for patients with biochemical recurrence or for metastatic disease, often consists of androgen deprivation therapy (ADT) with a gonadotropin hormone-releasing hormone (GnRH) agonist or by surgical castration. About half of patients with prostate cancer is estimated to receive ADT at some point during the course of their disease. 6 While most patients will respond initially to ADT with corresponding improvements in biochemical, clinical or radiographic parameters, most patients eventually experience progression despite castrate testosterone levels, and thus a need for new, ongoing treatments are necessary. To this end, multiple treatment modalities have been explored since the landmark trials that led to the approval of docetaxel for metastatic castration-resistant prostate cancer (CRPC).7,8
This review summarizes pre-clinical and clinical data that led to the Food and Drug Administration (FDA) approval of several new agents in the treatment of prostate cancer and discusses mechanisms of action and rationale for the use of these drugs in the pre- or post-docetaxel-refractory disease setting (see Table 1 for summary). This review serves to raise awareness of newly available drugs, some of which have shown overall survival benefit, but unanimously have changed the landscape of treatment in advanced prostate cancer.
New pharmacotherapies for Prostate cancer.
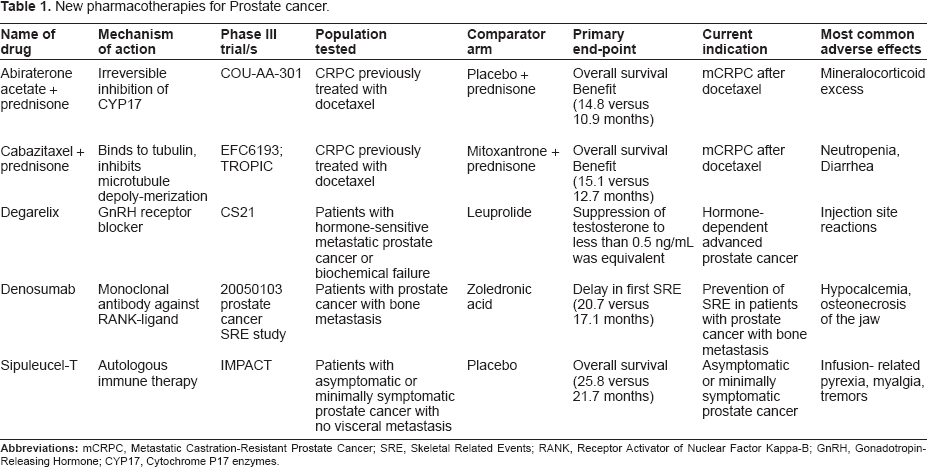
Mechanisms of Castration Resistance
ADT has been the cornerstone of treatment for locally advanced and metastatic prostate cancer, which can be achieved either through chemical castration with Gonadotropin Releasing Hormone Agonists (GnRH-A) and antagonists or by surgical castration through orchiectomy. Monotherapy with the use of nonsteroidal anti-androgens have also been used on occasion, as well as in conjunction with ADT, termed combined androgen blockade (CAB) or total androgen blockade. Although the use of CAB offers survival advantage of about 2%–3%, 9 it comes at the expense of increased cost and toxicity. While most patients will respond to ADT initially, this occurs at varying durability with eventual castration resistance gradually ensuing. Therefore, there is an increasing need to determine ways of abrogating the phenomenon of castration resistance. The primary use of ADT is commonly referred to as first line use of hormonal therapy. When castration resistance emerges, further hormonal manipulation beginning with anti-androgen withdrawal is often employed as a first step. Henceforth, second-line hormonal manipulation is undertaken to achieve additional PSA responses.
CRPC is characterized by a progressive rise in PSA despite castrate levels of testosterone, which typically is <50 ng/dL though emerging studies have shown that <20 ng/dL may be more ideal.10,11 Biologically, the androgen receptor (AR) plays a key role in the mechanisms that underlie the progression of hormone-dependent prostate cancer to one of castration resistance. Although ADT reliably decreases serum testosterone in >90% of cases, the levels of intratumoral decline in androgen may be less so.12,13 Therefore, this allows expression of AR and other androgen-responsive genes unchanged. Furthermore, it had been shown that the AR is persistently upregulated during progression in certain prostate cancer cell lines. 14 Various pathways, both androgen-dependent and independent, have been described in bringing about castration resistance. 15 Several involve the AR signaling pathway which includes de novo or intratumor-derived androgens, 16 which signal and activate the AR since intratumoral conversion of weak adrenal androgens dihydroepiandrosterone (DHEA) and androstenedione into potent testosterone and dihydrotestosterone can occur, effecting AR activation. 17 Other androgen-dependent mechanisms including AR overexpression or amplification, AR upregulation at the transcriptional levels, change in co-factor levels, and promiscuity of the AR in binding with other ligands can also occur. 7 Still others involve bypassing usual known routes of testosterone conversion to dihydrotestosterone. 18 Androgen-independent mechanisms bypassing the AR include phosphatase and tensin homologue (PTEN) via Akt-activation. 19
The pathways operative in the progression of castration resistance is essential to the understanding of drug development in the treatments especially of metastatic CRPC. The following section discusses the most recent therapies currently approved by the US FDA for the treatment of prostate cancer.
Recent Agents Approved for prostate cancer
Degarelix
Background on degarelix
While gonadotropin receptor hormone (GnRH) agonists are the most widely used forms of ADT, 20 they do have certain drawbacks, the most important of which is the transient surge in serum testosterone that has the potential to stimulate tumor growth, as well as causing a variety of effects including worsening urinary symptoms, bony metastasis leading to bone pain or cord compression. 21 The initial testosterone surge is blocked by concomitant anti-androgen use during the first few weeks of treatment, which is also not without side-effects. This led to the development of a new class of ADT, the GnRH receptor antagonists. GnRH receptor blockers block the GnRH receptor directly in the pituitary, rapidly blocking the release of LH and FSH reducing testosterone secretion. Since those GnRH antagonists do not cause the initial stimulation of the LH production, they do not cause the testosterone surge or clinical flare. 22 Abarelix was the first GnRH receptor blocker developed for prostate cancer treatment but was associated with systemic allergic reactions. 23 Further development brought about degarelix, a GnRH receptor antagonist that was approved in December 2008 by the FDA in the US for the treatment of patients with advanced prostate cancer, followed in February 2009 by EMEA approval for adult males with advanced hormone- dependent prostate cancer. Degarelix obviates a lot of the systemic side-effects of other GnRH antagonists such as abarelix as it does not have the histamine- releasing potential effects. 24
Early clinical phase I and II studies
Degarelix is a fully synthetic, linear decapeptide amide analogue of natural GnRH that contains seven amino acids, five of which are D-amino acids. 25 An early phase I study showed safety in 36 healthy young male volunteers where degarelix was administered intravenously, intramuscularly or subcutaneously with all routes resulting in acute suppression of LH, FSH and testosterone, 26 but high concentrations were achieved after intramuscular or subcutaneous administration resulting in a longer half-life compared to the intravenous administration.
Several multicenter, open-label, randomized, dose-finding phase II studies were conducted to determine the optimal dose of degarelix. The first study CS02 involved 129 patients with locally advanced or metastatic prostate cancer with a PSA above 20 ng/mL. 27 Patients who received the 80/80/40 mg degarelix regimen (80 mg on day 0, 80 mg on day 3, 40 mg on day 28) benefited the most from suppression of testosterone at days 3, 28 as well as after 6 months, with rapid PSA declines. CS12 and CS14 were two open-label randomized, 1- year studies, of similar design comparing different dosing regimens. In the CS12 study, 189 patients were randomized to one of six degarelix treatment groups: initial dose of either 200 or 240 mg followed by monthly maintenance disease of 80, 120, or 160 mg. 92% of the patients who received a 240 mg starting dose of degarelix achieved a testosterone level < 0.5 ng/mL at day 3, compared to 88% of the patients who received the 200 mg and the difference was maintained at 1 month (95% versus 86%; P = 0.048). The maintenance dose of 160 mg provided the highest efficacy in terms of providing long-term testosterone suppression. 28 The CS 14 study demonstrated that 89% of the 127 patients achieved testosterone levels of <0.5 ng/mL by day 3, with improved testosterone control after one month of treatment. Furthermore, the maintenance dose of 80 mg appeared the most effective in maintaining castrate testosterone levels to 1 year (98% for 80 mg, 93% for 60 mg). 29 The median time to a 50% reduction of PSA was 14 days and the median time to 90% reduction was 56 days in both maintenance dose regimens.
Pivotal phase III study
Given the promising phase II trials using degarelix, a phase III three–armed randomized trial (CS21) evaluating the efficacy and safety of two doses of degarelix versus a standard 7.5 mg dose of the LHRH agonist leuprolide was conducted, with the primary endpoint being suppression of testosterone to <0.5 ng/mL at all monthly measurements from day 18 to day 364. 30 In this trial, 610 patients with biochemical failure as well as those with hormone-sensitive metastatic disease were randomized into three treatment arms, namely degarelix at 240 mg initial dose followed by monthly subcutaneous doses of 80 mg, the 2nd arm with an initial 240 mg followed by monthly 160 mg doses and the 3rd arm with monthly 7.5 mg intramuscular injections of leuprolide, along with bicalutamide 50 mg daily for prevention of flares. The primary end point was achieved similarly in all arms, 97.2% of the patients in the first arm, 98.3% of the patients in the second arm and 96.4% of the patients in the third arm. PSA levels were also measured at days 14 and days 28 of initiation of treatment and were statistically significantly lower in the first two degarelix arms as compared with the leuprolide arm, with median PSA declining by 64% in the 240/160 arm, 65% in the 240/80 arm and 18% in the leuprolide arm. Exploratory post-hoc subgroup analyses performed on the CS21study showed that higher testosterone levels led to slower achievement of castrate levels though still significantly faster for the degarelix arm in all subgroups compared to the leuprolide group. 31
An extension trial involving 134 prostate cancer patients who were switched from leuprolide to degarelix at either a monthly maintenance dose of 80 mg or 160 mg showed serum testosterone of <0.5 ng/mL was sustained during the observation period from days 3–84. 32 PSA levels were reduced more rapidly during the first 28 days of treatment in the degarelix groups with median reduction of 65% at day 14 and 83% at day 28 whereas the leuprolide group had a median decrease in PSA of 20% at day 14 and 68% at day 28. There was a noted initial increase of FSH level in the leuprolide arm, followed by a decrease of about 75% of the FSH level at day 28, with potential significance in the transient surge in FSH in the leuprolide arm given the role of FSH as a driver of bone loss because of bone resorption. Thus, stronger suppression of FSH by the antagonist suggests a potential advantage which could be addressed in studies comparing the BMD in treatment groups. 33
Safety
Degarelix was found to be well tolerated in all phases of the clinical trials. The most frequent adverse events in the phase II studies were typical androgen deprivation symptoms that included hot flushes, fatigue, as well as injection-site pain (37%, 11% and 9%, respectively). No drug-related deaths occurred.28,31 Degarelix injection induced a higher rate of injection site reactions than leuprolide (40% versus 1%, P < 0.001) although the local reactions occurred mostly after the first injection and degarelix induced more chills (4% versus none in the leuprolide arm) which generally occurred 5–10 hours after administration. In addition, cardiovascular effects on the use of both drugs were almost similar, including QT prolongation, arrhythmias and ischemic heart disease, all occurring <10% of the cases. 34
Sipuleucel-T
Background
Sipuleucel-T (APC 8015, Provenge™, Dendreon) is an active cellular immunotherapy, a therapeutic cancer vaccine that is designed to stimulate an immune response to prostate cancer. It consists of autologous peripheral blood mononuclear cells (PBMCs), obtained through leukapharesis and cultured in vitro for 3 days with PA 2024, a prostatic acid phosphatase (PAP) fusion protein and granulocyte-macrophage colony-stimulating factor (GM-CSF). These cells are then reinfused into the patient inducing an immune response to PAP-expressing prostate cancer cells. 35 Sipuleucel-T was the first of its kind anti-cancer immunotherapy agent to be approved by the US FDA in April 2010, indicated for treatment of patients with asymptomatic or minimally symptomatic metastatic CRPC.
Early phase I and II studies
The clinical development of sipuleucel-T was based on preclinical demonstration of lymphocytic infiltrates in rat prostate tissue following administration of antigen presenting cells (APCs) incubated with a fusion protein of rat prostatic acid phosphatase (PAP) linked to rat granulocyte-macrophage colony-stimulating factor (GM-CSF). 36 In the human studies of sipuleucel-T, prostate cancer patients treated with PBMCs containing APCs incubated with PA2024, the recombinant fusion protein of human PAP-GM-CSF, an immune response was demonstrated, there was no dose limiting toxicity and a PSA response was observed in 10%–15% of the patients.37,38
Phase III studies
Two initial phase III trials were conducted to study the efficacy of sipuleucel-T in patients with progressive metastatic CRPC. The D9901 phase III placebocontrolled trial included 127 patients with progressive metastatic CRPC that were randomly assigned in a 2:1 ratio to be treated with Sipuleucel-T versus placebo every two weeks. 39 The study failed to meet its primary endpoint of median time to disease progression (11.7 weeks for Sipuleucel-T vs. 10.0 weeks for placebo; P = 0.052). However, median overall survival (OS) was significantly improved by 4.5 months in favor of Sipuleucel-T (25.9 months vs. 21.4 months, P = 0.01). Meanwhile, D9902A was a double-blind, placebo controlled trial of patients with metastatic, asymptomatic, CRPC in which the study halted after 98 patients were enrolled given “negative” findings of the D9901 study. In the combined analysis from D9901 and D9902A however, a significant improvement in OS was found in the sipuleucel-T group versus placebo (23.2 months vs 18.9 months, P = 0.01), though no improvement in median time to progression (TTP) was noted (11.7 vs. 9.7 weeks, P = 0.111), 40 which were the predefined primary endpoint. 41 Given the OS benefit, and after further discussion with the FDA, another phase III study called the IMPACT (Immunotherapy for Prostate AdenoCarcinoma Treatment trial) was launched which ultimately led to the landmark trial that got Sipuleucel-T approved (D9902B). 42 In this double-blind placebo-controlled multicenter study, 512 patients with asymptomatic or minimally symptomatic metastatic CRPC patients with no visceral metastases were randomized to Sipuleucel-T (n = 341) versus placebo (n = 171) in a 2:1 ratio. Treatment was given as over 1 hour infusion every 2 weeks at weeks 0, 2 and 4. The vast majority of the patients were chemo-naïve with <10% having received prior chemotherapy and were required to be off steroids. The patients who were pre-treated were required to be off chemotherapy for more than 3 months. The primary end point in this study was met with a median OS for the treatment arm improved by 4.1 months (25.8 months vs. 21.7 months, P = 0.03), after a median follow-up of 34.1 months. A relative reduction of 22.5% in the risk of death compared with placebo was reported in the Sipuleucel group (P = 0.03). The benefit was seen across groups regardless of baseline PSA levels, lactate dehydrogenase, alkaline phosphatase, number of bone metastases, Gleason score, ECOG performance status, and severity of pain. Similar to the preceding trials however, the secondary endpoints of time to objective disease progression, showed no statistical significance.
Safety
Sipuleucel-T was generally well tolerated in all clinical trials. The most common toxicities were infusion-related rigors, pyrexia, tremors, chills, myalgia, hypertension, hyperhydrosis and groin pain. These events were generally mild or moderate in severity and generally resolved within 1 to 2 days which led to hardly any treatment interruption occurring in only 0.9% of the patients.
Cabazitaxel
Background
Cabazitaxel (Jevtana™; Sanofi-Aventis, Paris, France) is a semi-synthetic taxane that uses a precursor molecule extracted from yew tree needles. Cabazitaxel inhibits microtubule depolymerization and cell division by binding to tubulin, resulting in cell cycle arrest. It was selected for clinical development due to its poor affinity for ATP-dependent drug efflux pump P-glycoprotein (P-gp) 43 and its greater blood-brain barrier penetration, 44 compared to paclitaxel and docetaxel in preclinical models. Cabazitaxel has also demonstrated superior in vitro cytotoxicity compared to docetaxel in several murine and human cancer cell lines. 43 Cabazitaxel was approved by the US FDA on June 17, 2010 for patients who have failed docetaxel as second-line therapy.
Early phase I and II studies
An early phase I clinical trial showed safety in 25 patients with advanced solid malignancies refractory to conventional treatments, the largest subgroup comprising of prostate cancer patients (8 patients, 32%), were treated with 102 courses of 3-weekly cabazitaxel at 4 dose levels, ranging from 10 mg/m2 to 25 mg/m2. 43 A total of 22 patients had received prior chemotherapy (88%), and 8 patients had received prior taxane-based therapy (32%). A median of 4 cycles (range 1–9) was administered. Pharmacokinetic analyses showed a rapid initial phase was followed by a longer intermediate phase (t1/2 = 2.5 minutes and 1.3 hours, respectively) with a prolonged terminal phase (t1/2 = 77.3 hours). The dose-limiting toxicity (DLT) of cabazitaxel was neutropenia, with 1 case of febrile neutropenia and 2 cases of grade 4 neutropenia occurring at a dose of 25 mg/m2 without routine use of growth factor support in these studies, although ultimately administered in patients developing grade 4 neutropenia. Non-hematologic toxicities were generally mild in nature; the most commonly encountered adverse events were diarrhea (52%), nausea (40%), and vomiting (15%). Objective responses in this study included 2 confirmed partial responses observed in 2 patients with prostate cancer, with minor responses seen in two patients, one of whom had prostate cancer. Twelve patients (48%) had stable disease for greater than 4 months.
The clinical approval of cabazitaxel follows one of the fastest tracks of clinical drug development with no phase II trials of cabazitaxel in patients with advanced prostate cancer ever conducted, but a phase II study in 71 patients (61 evaluable) with metastatic breast cancer was central to the 25 mg/m2 dosing selected for the eventual phase III trial. 45 After a median follow-up of 20 months, the median overall survival was 12.3 months and median time to progression was 2.7 months.
Pivotal phase III trial
Given the promising results in the phase I trial of cabazitaxel, a randomized, multicenter, multinational, phase 3 trial (EFC6193; TROPIC) was conducted to compare cabazitaxel with mitoxantrone in patients with CRPC who had progressed despite docetaxel-based chemotherapy. 46
Patients were randomly assigned to receive either cabazitaxel 25 mg/m2 intravenously over 1 hour or mitoxantrone 12 mg/m2 intravenously over 15–30 minutes on day 1 of each 21-day cycle, with prednisone 10 mg daily. Premedication, consisting of single intravenous doses of an antihistamine, corticosteroid and histamine H2-antagonist, was administered 30 minutes prior to cabazitaxel administration, while anti-emetic prophylaxis was given at physicians’ discretion. Due to the risk of mitoxantrone-induced cardiotoxicity, a maximum of 10 cycles of treatment were allowed. Patients were given prophylactic granulocyte colony stimulating factor only if prolonged neutropenia was encountered in earlier cycles. The primary endpoint of the study was overall survival. Seven hundred and fifty-five patients were randomized, 371 of the patients in each arm received the intended treatment however more patients receiving cabazitaxel completed the full treatment course compared to patients receiving mitoxantrone. The protocol was amended to exclude patients previously treated with a cumulative docetaxel dose lower than 225 mg/m2 to increase the likelihood of enrolling a true “docetaxel-refractory” population. The baseline characteristics and treatment history of the two groups were similar.
At median follow-up of 12.8 months, an overall survival benefit was demonstrated for patients receiving cabazitaxel compared with mitoxantrone (15.1 month vs. 12.7 months, HR 0.70, P < 0.0001). Secondary outcomes that also favored treatment with cabazitaxel over mitoxantrone included progression-free survival (2.8 vs. 1.4 months, HR 0.74, P < 0.0001), tumor response (14.4% vs. 4.4%, P = 0.0005), PSA response (39.2% vs. 17.8%, P = 0.0002), time to tumor progression (8.8 vs. 5.4 months, P, 0.0001), and time to PSA progression (6.4 vs. 3.1 months, P = 0.001). However, pain control and time to pain progression were similar among the two treatment arms.
Safety
As was shown in the phase I experience, the most common toxicity associated with cabazitaxel therapy in the TROPIC trial was neutropenia (94%). 46 Grade ≥ 3 neutropenia occurred in 82% of cabazitaxel patients, with 8% of patients developing febrile neutropenia. Furthermore, the incidence of neutropenia varied significantly by region, with rates of neutropenia in North America exceeding those in the Europe, therefore prompting the prophylactic use of growth factors especially in those at high risk for neutropenia, such as those patients who are 65 years or older, have poor performance status, had previous episodes of febrile neutropenia, extensive prior radiation ports, poor nutritional status, or other serious comorbidities, with further approval of cabazitaxel in the US. 47 Common all grades non-hematologic toxicities in patients receiving cabazitaxel included diarrhea (47%), fatigue (37%), and asthenias (20%). A total of 18 patients (5%) died within 30 days of the last cabazitaxel infusion, compared with 9 patients (2%) receiving mitoxantrone therapy. In the cabazitaxel arm, 7 patients (2%) died of complications related to neutropenia, while 5 patients (1%) died of cardiac causes. The rates of neuropathy with cabazitaxel were relatively low, only 1% of patients reporting a grade 3/4 event (14% for all grades) though patients with severe neuropathy post-docetaxel were excluded from TROPIC.
Denosumab
Background
Bone metastasis frequently occurs in prostate cancer patients and may result in increased morbidity and mortality. Furthermore, bone pain, pathological fractures, hypercalcemia, and other bone-related complications undermine quality of life. When cancer cells metastasize to the bone, their growth is promoted under the influence of a variety of growth factors that are supplied from the bone as a consequence of osteoclastic bone resorption. 48 This, in turn, causes an increased production of osteoclast- and osteoblast-stimulating cytokines in these cancer cells, leading to a further increase in bone remodeling. This vicious cycle between bone and metastatic cancer cells supports the development and progression of bone metastases. Accumulating evidence shows that receptor-activator of NF Kappa-B ligand (RANKL), a cytokine expressed in osteoblasts and bone marrow stromal cells, plays an important role in the formation, activation, and survival of osteoclasts, which are key players in bone remodeling, and thus has been used in in men with prostate cancer receiving hormone therapy to prevent bone loss.49,50
Denosumab, a fully human monoclonal anti-RANKL antibody, serves to inhibit and disrupt the ongoing aforementioned vicious cycle. The etiology of compromised bone heath in castrate resistant prostate cancer are multifold and encompasses reduced bone mineral density from castration treatment, accompanying glucocorticoid use, and inherent biologic changes related to the cancer and bone metastases. 51
Denosumab was approved by the FDA in November 2010 for the prevention of skeletal-related events in patients with bone metastases from solid tumors. It differs from zoledronic acid in that denosumab is administered subcutaneously with less concern for renal toxicity or for renal dose adjustments. 52
Early phase II trials
A phase II study looked at the effect of denosumab on elevated urinary N-telopeptide, which represents excessive bone resorption, in 111 patients with mainly prostate or breast cancer who had bone metastases. 53 Randomization to bisphosphonate therapy (pamidronate or zoledronic acid) or subcutaneous denosumab on a fixed schedule was performed. Results showed that the proportion of patients with urinary N-telopeptide lower than 50 was maintained at week 25 in 64% of the denosumab arm compared to 37% in the bisphosphonate arm. Skeletal-related events was notably lower in the denosumab group (8%) versus 17% in the bisphosphonate arm. There was no difference in the rates of adverse events.
Pivotal phase III trial
Denosumab was initially studied in men with non-metastatic prostate cancer receiving ADT. In a double-blinded phase III trial, 1,468 men received either denosumab (60 mg) subcutaneously every six months or placebo. At 24 months, bone mineral density of the lumbar spine had increased by 5.6% in the denosumab group compared with a loss of 1.0% in the placebo group (P value of <0.001). There was also a decrease in incidence of new vertebral fractures at 36 months for patients on denosumab (1.5% versus 3.9%, P value of 0.006). Rates of adverse events were similar between the two groups in the trial. 49 This trial has led to the recent FDA approval for a new indication of denosumab increase bone mass in men at high risk for fractures while receiving ADT.
In the second study, denosumab was compared to zoledronic acid in 1904 patients with bone metastases from castrate-resistant prostate cancer. 54 While there were no significant effects on PSA progression or overall survival, denosumab significantly extended the time to first on-study skeletal-related event which was the primary endpoint, with a median time of 20.7 months compared with 17.1 months for zoledronic acid (hazard ratio of 0.82, P value of 0.008 for superiority).
Safety
Common side-effects with the use of denosumab included hypocalcemia with an incidence of 13% (versus 6% in the zoledronic acid arm). 54 Therefore, sufficient calcium and vitamin D levels must be reached and maintained before and during denosumab therapy. Other common side-effects included urinary and respiratory tract infections, cataracts, constipation, rashes, and joint pain. A small study found a slightly increased risk of cancer and severe infections, but these results did not reach statistical significance. 55 It has been proposed that the increase in infectious complications with denosumab use might be related to the role of RANKL in the immune system, which is expressed by T helper cells and thought to be involved in dendritic cell maturation. 56 Osteonecrosis of the jaw occurred infrequently but was found to be increased in patients receiving denosumab (2% of the denosumab group compared with 1% of the zoledronic acid group), although exact mechanisms are yet unclear. 52
Abiraterone
Background
Abiraterone acetate (Zytiga™, Jansenn Biotech, Inc.) is a novel inhibitor of CYP17, an enzyme responsible for steroid biosynthesis leading to production of androgenic and estrogenic steroids. Given persistence of ligand-mediated androgen receptor signaling as one of the key drivers of castration resistance, the design of a potent, selective, and irreversible inhibitor of CYP17 was formulated. Abiraterone (CB7598) is an oral, potent, selective, and irreversible inhibitor of CYP17 that is 10- to 30-fold more potent than the non-selective inhibitor, ketoconazole. Abiraterone acetate (AA) along with prednisone use attained approval from the US FDA on April 28, 2011 for metastatic CRPC after docetaxel failure. 57
Early phase I and II studies
The prodrug 3-β-O-acetate (AA, CB7630) is rapidly de-acetylated to the active metabolite in vivo, in contrast to its parent drug which has poor bioavailability. 58 AA is highly protein bound and is metabolized in the liver by CYP3 A4 and SULT2 A1 to inactive metabolites, excreted in feces (~88%) and urine (~5%) with a terminal half- life of 12 ± 5 hours. In preclinical toxicology studies, AA reduced the weights of androgen dependent organs and had minimal side effects in other organs. 59 Earlier first-in-man studies established suppression of testosterone synthesis in non-castrate patients for 12 days continuously to men with prostate cancer. 60 A phase I trial included 21 chemotherapy-naïve men with CRPC and was treated with once-daily, continuous AA, which escalated through five doses (250 to 2,000 mg) in three-patient cohorts. 61 Although antitumor activity was observed at all doses, the 1,000 mg dose was selected for cohort expansion (n = 9) because of a plateau in the pharmacodynamic effect. Declines in prostate-specific antigen ≥ 30%, 50%, and 90% were observed in 14 (66%), 12 (57%), and 6 (29%) patients, respectively, and lasted between 69 to ≥578 days. To rapidly evaluate the antitumor activity of abiraterone acetate, expansion of the phase I study into a two-stage, single-arm, phase II trial that included 54 chemotherapy-naïve CRPC men with further expansion of 42 patients, treated with once daily 1,000 mg abiraterone acetate using a two-stage design to reject the null hypothesis if >7 patients had a PSA decline of ≥50% was undertaken. 62 Evaluation every 12 weeks and circulating tumor cell (CTC) enumeration were performed. A decline in PSA of ≥50% was observed in 28 (67%) of 42 phase II patients, and declines of $90% were observed in eight (19%) of 42 patients. Partial responses (Response Evaluation Criteria in Solid Tumors) in nine (37.5%) of 24 phase II patients with measurable disease were seen, along with declines in CTC counts. The median time to PSA progression (TTPP) on AA alone for all phase II patients was 225 days (95% CI, 162 to 287 days). Exploratory analyses performed showed that the addition of dexamethasone at disease progression reversed resistance in 33% of patients regardless of prior treatment with dexamethasone.
AA was also tested in phase II clinical trials in docetaxel-pretreated CRPC.63,64 The first study was conducted in 58 metastatic CRPC patients who experienced treatment failure with docetaxel-based chemotherapy and received AA (1,000 mg daily) with prednisone (5 mg twice daily). Twenty-seven (47%) patients had received prior ketoconazole. The primary outcome of ≥50% decline in PSA was confirmed in 22 (36%) patients, including 14 (45%) of 31 ketoconazole-naïve and seven (26%) of 27 ketoconazole-pretreated patients. In 22 patients with RECIST-evaluable lesions, partial responses were seen in four (18%) of patients. Furthermore, improved performance status was seen in 28% of patients. Median time to PSA progression was 169 days (95% CI, 82 to 200 days). CTC conversions with treatment from ≥5 to <5 were noted in 10 (34%) of 29 patients. The majority of AA-related adverse events were grade 1 to 2, and no AA-related grade 4 events seen, with reduction of incidence of mineralocorticoid-related by adding low-dose prednisone. 63 Another phase II multicenter study confirmed the significant antitumor activity of daily 1,000 mg AA given in patients with docetaxel-pretreated CRPC. 64 The primary outcome of ≥50% PSA decline in at least 20% patients was met with 47 patients enrolled. Results have shown a PSA decline of ≥30%, ≥50% and ≥90% in 68%, 51%, and 15% of patients, respectively. Median time to PSA progression was 169 days (95% CI, 113 to 281 days). CTCs were enumerated in 34 patients; 27 (79%) of 34 patients had at least five CTCs at baseline. Eleven (41%) of 27 patients had a decline from at least five to less than 5 CTCs, and 18 (67%) of 27 had a ≥30% decline in CTCs after starting treatment with AA. Mineralocorticoid excess was effectively managed with a mineralocorticoid receptor antagonist or a low-dose of glucocorticoid.
Pivotal phase III trial
Given the promising results in the phase I and II trials of abiraterone, a phase III trial (COU-AA-301) that ultimately led to the FDA-approval of abiraterone acetate for metastatic CRPC after progressing through docetaxel enrolled 1195 patients who were randomized in a 2:1 ratio to receive 5 mg of prednisone twice daily with either 1000 mg of abiraterone acetate (797 patients) or placebo (398 patients). 65 The trial was conducted to compare the safety and efficacy of AA in men with mCRPC previously treated with docetaxel, with the primary endpoint of OS.
After a median follow-up of 12.8 months, the overall survival was longer in the abiraterone acetate–prednisone group than in the placebo–prednisone group (14.8 months vs. 10.9 months; HR 0.65; P < 0.001). Since these results exceeded the preplanned criteria for study termination, data were unblinded at the interim analysis. All secondary end points, including time to PSA progression (10.2 vs. 6.6 months; P < 0.001), progression-free survival (5.6 months vs. 3.6 months; P < 0.001), and PSA response rate (29% vs. 6%, P < 0.001), favored the treatment group.
Safety
The safety profile observed for patients who received AA was fairly similar with the placebo group with most adverse events occurring as grade 1 and 2 toxicity, with fatigue as the most common, followed by back pain, nausea, constipation, bone pain, and arthralgias. Grade 1 and 2 urinary tract infection was more frequent in the abiraterone acetate group (12%, vs. 7% in the placebo group; P = 0.02). Similar rates of treatment discontinuation due to adverse events were seen in both groups, 19% for AA and 23% for placebo. Given the CYP17 inhibition, adverse events associated with elevated mineralocorticoid levels were expectedly more common in the AA arm compared with placebo (fluid retention and edema, hypokalemia, and hypertension, cardiac disorders and liver-function test abnormalities were more common in the abiraterone acetate group than in the placebo group (55% vs. 43%, P < 0.001). No individual grade 4 adverse events occurred in 2% or more of patients in either treatment group.
Other promising agents in development
MDV3100
MDV3100 is an experimental androgen receptor antagonist drug that was developed by Medivation for the treatment of CRPC. MDV3100 has about a fivefold higher binding affinity for the AR compared to bicalutamide and does not promote translocation of AR to the nucleus and in addition prevents binding of AR to DNA and coactivator proteins. 66 In vitro LNCaP cells treated with MDV3100 showed expression of androgen dependent genes PSA and TMPRSS2 which were down-regulated in contrast to bicalutamide where the expression was up-regulated. MDV3100 also induces tumor cell apoptosis, and has no agonist activity.
In a phase I-II study, 140 patients with metastatic CRPC underwent dose escalations of MDV3100, starting at 30 mg daily. 67 The results demonstrated anti-tumor effects at all doses, including decreases in serum prostate-specific antigen of 50% or more in 56% of the patients studied. There were also responses in soft tissue in 22% of patients (13 out of 59) and conversion from unfavorable (≥ 5 CTC/7.5 ml of blood) to favorable (<5 CTC/7.5 ml of blood) circulating tumor cell counts in 49% of patients (25 out of 51). The median time for radiological progression was 47 weeks. The maximum tolerated dose of MDV3100 was 240 mg daily with the most common adverse event being dose-dependent fatigue (observed in 11% of patients). Long-term follow-up efficacy data from this trial was presented at ASCO-GU in February 2011, which showed MDV 3100 continues to demonstrate durable antitumor activity as evaluated by median times to PSA progression and radiographic progression.
An international, multi-center, randomized, placebo-controlled, phase III study (AFFIRM) has finished enrollment with a target accrual goal of 1199 patients to determine whether MDV3100 will detect an overall survival difference in patients with metastatic castration-resistant prostate cancer who were treated with prior docetaxel. A phase III trial (PREVAIL) looking at the use of MDV3100 in a chemotherapy-naïve population who have failed androgen deprivation therapy is currently ongoing.
In addition, there is an open phase II trial (TERRAIN) comparing MDV31000 with bicalutamide in prostate cancer patients who have progressed while on leuprolide or underwent surgical castration. The primary endpoint of the trial is progression-free survival. Another phase II trial that is examining the effects of MDV3100 monotherapy on prostate cancer patients who have not had any prior hormonal therapies or chemotherapy is open with target accrual of approximately 60 patients in Europe with PSA as the primary endpoint.
Cabozantinib
Cabozantinib (XL184), which is developed by Exelixis, is an inhibitor of the cell-signaling molecules hepatocyte growth factor receptor (MET) and vascular endothelial growth factor receptor 2 (VEGFR2). 68 MET signaling contributes to the spread of cancer cells while VEGFR2 plays a role in tumor blood vessel growth.
A phase II randomized discontinuation trial using cabozantinib in patients with metastatic CRPC with a target accrual of 200 patients but halted with 168 patients when high rates of clinical activity was observed, used cabozantinib 100 mg daily over 12 weeks with response assessed every six weeks. 69 Of the 100 evaluable patients in the lead-in phase, 86% (56/65) of patients in the trial had complete or partial resolution of lesions on bone scan as early as week 6. Improvement of pain was seen with 64% having improved pain and 46% had decreased or halted narcotics. There was improvement in anemia with a median rise of 2.2 g/dL in hemoglobin. Objective response in soft tissue was seen in 84% of patients. At week 12, the disease control rate was 71%, and the randomization was halted. The most common related adverse events were fatigue (11%), hypertension (7%), and hand-foot syndrome (5%) with drug discontinuation done in 10% of patients. This agent therefore shows promising results especially in terms of pain control and bone scan results and further investigation in phase III trials is anticipated.
TAK-700
TAK-700 (Orteronel, developed by Millennium Pharmaceuticals), is a novel, non-steroidal androgen synthesis inhibitor of the enzyme CYP17A1. In a phase I/II trial, 26 patients with metastatic CRPC were enrolled at five different dose levels ranging from 100 mg to 600 mg twice daily dosing. 70 All patients treated with ≥300 mg had a PSA decline. Twelve of 15 patients (80%) who received TAK-700 ≥ 300 mg for ≥3 cycles had PSA reductions of ≥50% and 4 of 15 (27%) had reductions ≥ 90%. The most common adverse events were fatigue followed by non-dose-related GI events such as nausea, constipation, and vomiting. Updated results on 96 patients was presented showing PSA response rates (≥50% decrease) at 12 weeks of 63%, 52%, 41%, and 62% in the 300 mg twice daily, 400 and 600 mg daily with prednisone, and 600 mg daily groups, respectively. 71 Of the 43 patients with RECIST-evaluable disease, 6had a partial response, 23 had stable disease, and 9 had disease progression. The mean circulating tumor cell numbers were also found to be decreased in all groups.
Other ongoing trials include a phase 1/2 study of TAK-700 in combination with docetaxel and prednisone in men with metastatic CRPC and two phase III trials comparing TAK-700 with prednisone versus placebo with prednisone in patients who are chemotherapy-naïve and post-docetaxel treatment.
Sequencing of treatment
While all the recent developments have brought about exciting changes to the landscape of prostate cancer therapy, clear guidance on how to sequence each of these drugs is unknown. The prevailing thought is to try and endeavor to have most patients see through each treatment in varying states of their disease. While treatment algorithms are not currently available, certain principles of therapy can be made in the treatment of advanced prostate cancer in the setting of these newly available drugs. 72 Long before any treatment existed for metastatic CRPC except for docetaxel, the debate had centered on when to commence chemotherapy and most clinicians commence chemotherapy in the symptomatic metastatic state, a practice that some have questioned, 73 considering the potential improvement in survival even in those who are minimally or asymptomatic patients. 74 While some guidelines have been put forth by European agencies, 75 suggesting commencement of chemotherapy in those who have failed through 2 prior hormonal manipulations, no such recommendations are available in the US. The practice of commencing chemotherapy in symptomatic patients has largely been adopted secondary to the belief that no alternative effective second-line therapy was available. However, subgroup analysis of the TAX-327 trial did indicate that patients who were asymptomatic at the time of receipt of docetaxel fared better than those who were symptomatic, with prolonged survival at a median of 25.6 months compared with those who were symptomatic with a median of 17.1 months (P = 0.009). 76 Whether or not these asymptomatic men would have done well regardless of receipt of treatment is unknown. Heretofore is the population of men for which vaccine therapy with Sipuleucel-T is currently indicated. Earlier studies with the use of vaccines in the post-chemotherapy setting have not been successful, and clinical studies that follow suggest that the use of immunotherapy has the greatest yield in earlier settings where tumor burden is lower and generation of immune responses against prostate cancer tumor-associated antigens is possible. 77 To this end, the prevailing thought is to initiate immunotherapy earlier on in the disease course, in asymptomatic or minimally symptomatic men with metastatic CRPC, the current indication for Sipuleucel-T (see Fig 1). While overall survival is significantly impacted, progression-free survival is not, suggesting that other mechanisms are operative in bringing about this change and perhaps an alteration in the biologic disease itself is causative in the improvement in survival. Nonetheless, this approach is pragmatic, since patients who are asymptomatic or minimally symptomatic do not have manifestations that urgently need to be addressed and certain approved therapies, such as docetaxel and abiraterone in particular, 76 have known pain palliative effects. How to best sequence agents in post-docetaxel chemotherapy are yet unknown. Current practice dictates that patients who have overall good performance status cycle through the currently approved second-line treatments of abiraterone or cabazitaxel, though individualized discussion of both merits and adverse effect profiles of each agent must be taken into consideration with each patient. With regard to bone-targeting agents, increasing awareness of the use of denosumab as front-line treatment for this population of patients is being implemented likely because of the ease of administration and less rigor in the kidney function monitoring. 52 In addition, promising radiopharmaceuticals such as samarium-153 lexidronam, are currently in use,78–80 and newer agents such as radium-223 have also shown promising results.81,82
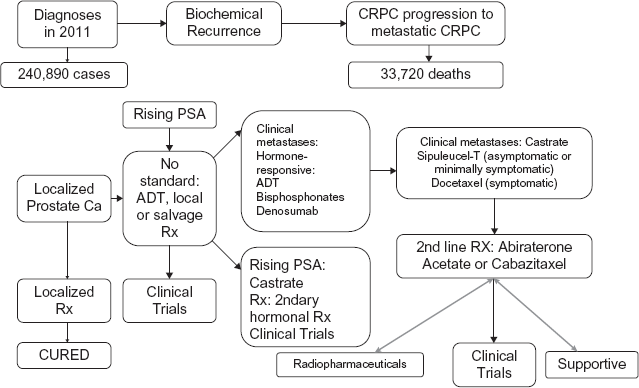
Natural history and changing landscape in the treatment paradigm for prostate cancer. Localized prostate cancer is treated with varying local therapies including surgery or radiation and majority are cured. About 30% will present with biochemical recurrence for which no current standard treatment (Rx) exists but ADT (Androgen deprivation therapy), further salvage options are often employed. Persistent PSA rise to castrate-resistant levels in the absence of metastases often results in use of secondary hormonal treatment (Rx). Progression to clinical metastases entails use of ADT and use of bone-targeting agents. However, with further CRPC (Castration-resistant prostate cancer), Sipuleucel-T is indicated for the asymptomatic or minimally symptomatic patient and docetaxel for symptomatic disease. Further standard 2nd line therapies approved in the US includes cabazitaxel and abiraterone.
Conclusions
The changing landscape in the treatment of prostate cancer has never been as robust. However, more work still needs to be done in improving the overall state of metastatic CRPC treatment, from aspects of increased surveillance and diagnostic measures of metastatic disease, utilization of optimal markers in determining when to begin therapy or when to switch, and to actual discovery of novel pathways and mechanisms of disease and the means to target them. Current clinical trial design approaches may have to be revisited, assessment of composite response to treatment as well as ascertainment of survival response (ie, progression-free survival versus overall survival) depending on the agent used, would be of paramount importance given several agents now that have shown overall survival gains. Certainly, moving these agents earlier in the clinical disease state is also being implemented, and currently a moving target.
Disclosures
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributor-ship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.
Conflicts of Interest
Dr. Aragon-Ching has served on the Speakers’ Bureau of Sanofi-Aventis and Centocor-Orthobiotech/Jansenn pharmaceuticals.