
Abstract
The treatment of exudative age-related macular degeneration (AMD) has been completely transformed by the development of drugs that bind vascular endothelial growth factor (VEGF). The antibody-based VEGF inhibitors bevacizumab and ranibizumab usually prevent the enlargement of choroidal neovascular membranes, reduce vascular permeability, and improve visual acuity. The newest VEGF inhibitor, aflibercept, is a soluble fusion protein that binds all isoforms of VEGF-A, VEGF-B, and placental growth factor with high affinity. Preclinical studies demonstrated aflibercept's ability to prevent experimental neovascularization and tumor growth in animal models. In phase 3 trials for exudative AMD, patients who received aflibercept avoided moderate vision loss and experienced improved visual acuity comparable to those who received ranibizumab. Additionally, patients who were treated with aflibercept 2 mg every 8 weeks (after 3 monthly loading doses) had similar visual results to those treated every 4 weeks. When treated as needed during the second year of the trials, patients were able to last an average of 3 months between aflibercept injections. Since its regulatory approval, aflibercept has also been found to perform well as a salvage therapy for eyes that respond incompletely to ranibizumab and bevacizumab. Because aflibercept can be administered less frequently than ranibizumab, it promises to decrease the frequency of patients’ visits to physicians’ offices in addition to the overall cost of AMD therapy.
Keywords
Introduction
Age-related macular degeneration (AMD) has challenged physicians and blinded an ever-increasing number of patients for many years. 1 AMD represents a compensatory angiogenic response by the choriocapillaris and an apoptotic response by the retinal pigment epithelium (RPE) to chronic ischemia and inflammation of the outer retina. Though dry or nonexudative AMD, which is characterized by areas of hyperplasia and atrophy of the RPE, constitutes the most common form of AMD, severe vision loss usually results from exudative AMD, 2 where choroidal neovascular membranes (CNVM) arise from the choroidal circulation, grow beneath the RPE or photoreceptors, chronically exude protein rich fluid, frequently bleed, and ultimately form fibrotic disciform scars. The resultant loss of central vision constitutes the leading cause of blindness in developed countries. 3 Physicians treated patients with various therapies, such as subcutaneous interferon, 4 surgical removal of the CNVM, 5 and macular translocation, 6 but all failed to slow vision loss in the majority of cases.
Laser photocoagulation that targeted extrafoveal and juxtafoveal CNVM became the first therapy to slow vision loss in selected patients. 7 Laser photocoagulation of subfoveal CNVM prevents enlargement of the lesions but also results in profound central scotomas. Ocular photodynamic therapy with intravenous verteporphin reduces vision loss by 50% in eyes with predominantly classic CNVM but rarely results in sustained gains in vision. 8 Though these therapies were embraced as therapeutic advances, ophthalmologists continued searching for treatments that would improve vision in most patients.
The hoped-for therapeutic advance for patients with exudative AMD emerged from an unlikely source–-oncologic research. Since cancers outgrow their blood supply once they reach a size of 2 mm3, Folkman (1971) opined that tumor growth requires an inducible angiogenesis factor. 9 In 1983, Senger discovered “vascular permeability factor” 10 and, in 1989, Ferrara and Connolly independently sequenced identical proteins–-the same as vascular permeability factor–-which became known as vascular endothelial growth factor (VEGF).11,12 Knowledge of VEGF biochemistry developed rapidly as did a growing appreciation for its role in both tumor growth and chorioretinal vascular diseases. Drugs capable of binding diffusible VEGF and preventing angiogenesis and vascular hyperpermeability were soon developed.
Developers of the first anti-VEGF drugs used two different technologies: aptamer technology to create pegaptanib and antibody technology to create bevacizumab and ranibizumab. Though results obtained from the treatment of exudative AMD with pegaptanib were somewhat disappointing, 13 bevacizumab and ranibizumab improve vision in the majority of patients.14–16 To create an even better VEGF-binding drug, scientists at Regeneron, Inc., (a growing biotechnology company) synthesized a soluble, fusion protein with native binding sequences from VEGF receptors. The resultant drug, aflibercept, underwent intensive testing, which led to its approval by the United States Food and Drug Administration (US FDA) on November 19, 2011, for the treatment of exudative AMD. This manuscript discusses the development of aflibercept, the rationale for its therapeutic use in patients with exudative AMD, and its performance in pivotal clinical trials.
Etiology of Macular Degeneration
The early signs of AMD often appear in the fifth and sixth decades of a patient's life, but the underlying biochemical changes begin during the first decade. Repeated visual transduction by the outer retina creates the highest metabolic demand of any tissue in the body. Rejuvenation of 11-cis retinol after it undergoes transformational changes by incident photons requires near continuous shedding and resynthesis of the photoreceptor segments outer discs.
Whereas the retinal circulation serves the inner two thirds of the retina, the outer neurosensory retina and pigment epithelium is nourished by the choriocapillaris, the dense capillary network of the inner choroid. Both micronutrients and macronutrients pass continuously from the choriocapillaris, through Bruch's membrane, to the RPE and photoreceptors. Plasma lipoproteins carry lipophilic nutrients such as carotenoids, 17 cholesterol, 18 and vitamin E to the RPE where they are removed and prepared for transport to the photoreceptors. Subretinal proteins such as docohexaenoic acid (DHA) are believed to rapidly transport unesterified cholesterol (UC) to and from the retina. 19 However, UC accumulates in the outer retina, and, since it is cytotoxic, it must be either metabolized or removed. Bruch's membrane lipoproteins are assembled from multiple lipids and UC is reesterified, thus creating the abundant cholesterol, which, along with fatty acids such as linoleate, accumulates in Bruch's membrane. 20
Thus the RPE appears to have 2 transport circulations: DHA apically to the photoreceptors and lipoproteins basally to the systemic circulation. 21 Lipoproteins probably traverse Bruch's membrane by diffusion and enter the choriocapillaris by transcytosis; however, the accumulation of esterified cholesterol in Bruch's increases resistance to diffusion, 22 resulting in decreased clearance of lipoproteins and an accumulation of lipids within Bruch's, just external to the basal lamina. Over time, these sequestered lipids are transformed into species such as linoleate hydroperoxide 23 and 7-ketocholesterol. 24 Unfortunately, lineolate hydroperoxide, which is also found in atherosclerotic plaque, 25 is chemotactic, proinflammatory, and cytotoxic. 21 The components of apolipoproteins are degraded by both oxidative and nonoxidative processes resulting in the formation of lipoprotein-derived debris, a major component of soft drusen. 26 In this manner, it appears that peroxidized lipids induce choroidal neovascularization by inciting chronic inflammation.27–29
The “oil spill” 21 in Bruch's membrane decreases diffusion of nutrients and metabolites in both directions. Decreased oxygen diffusion, coupled with increased metabolic demands due to lipid accumulation and induced inflammation, results in relative hypoxia of the RPE and photoreceptors. Low oxygen tension prevents enzymatic hydroxylation of hypoxia inducible factor (HIF) 1-α, the cell's “oxygen sensor,” thereby enabling it to dimerize with HIF 1-β. This stable complex binds to the promoter region of the VEGF gene and upregulates VEGF synthesis. 30 Several growth factors and inflammatory cytokines, including interleukin-1, interleukin-6, and several chemokines are upregulated by the chronic inflammation and ischemia, thereby promoting the growth of new blood vessel complexes from the choriocapillaris.
VEGF Biochemistry
VEGF is actually several closely related molecules that segregate into 6 families, VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor, each of which is responsible for regulating functions critical to angiogenesis, inflammation, and cellular survival. For example, VEGF-C and VEGF-D stimulate lymphangiogenesis, but most angiogenesis-related processes are regulated by isoforms of VEGF-A. 31 The VEGF-A family has at least 6 major and 8 minor isoforms. 32 The longer VEGF-A isoforms, VEGF183, VEGF189, and VEGF206, contain basic amino acid sequences that promote binding to cell membranes and sequestration within the intercellular matrix; VEGF145 and VEGF121 bind minimally to matrix proteoglycans and diffuse freely; and 50% to 70% of VEGF165, the isoform responsible for most human angiogenesis, 33 is matrix bound. 34 The longer, matrix-bound VEGF isoforms form a reservoir that can be activated by tissue injury to amplify the VEGF response. Diffusible VEGF activates matrix metalloproteinase-9, which dissolves the intercellular matrix and allows plasmin to cleave sequestered VEGF isoforms resulting in the release of VEGF110.33,34
VEGF binds to 3 transmembrane receptor molecules, VEGFR1 (flt-1), VEGFR2 (flk-1), and VEG-EFR3 (flt-4), expressed by vascular endothelial, retinal pigment epithelial, Mueller, and retinal glial cells.35,36 Activation of VEGFR3 by its ligands, VEGF-C and VEGF-D, stimulates lymphangiogenesis, whereas VEGFR2 regulates most of the processes associated with angiogenesis. Binding of a VEGF homodimer to VEGFR2 dimerizes the receptors and induces a transmembrane conformational change, which activates the receptors’ intracellular tyrosine kinase moieties. This upregulates several downstream biochemical pathways, including MAP/MEP/ERK, MAPK, and phosphotidylinositol 3-kinase (PI3k),37–40 which lead to endothelial cell proliferation, migration and swelling; attraction of monocytes, leukocytes, and endothelial cell progenitors; vascular dilation, 41 phosphorylation of junctional proteins and matrix metalloproteinases; 42 and improved survival of several cell types. Though VEGFR1 has a greater affinity for VEGF165 (KD = 10–30 pM) than does VEGFR2 (KD = 75–125 pM), its role in angiogenesis is unclear. Activation of VEGFR1 by VEGF-B promotes coronary vascular growth, but activation by VEGF-A attracts monocytes and leukocytes, thereby suggesting that VEGFR1 may act as a weak instigator of angiogenesis. Alternatively, VEGFR1 may act as a “decoy” receptor by strongly (KD = 45 pM) binding placental growth factor, thereby displacing VEGF165 and allowing it to preferably bind and activate VEGFR2. 34
Two molecular binding sites determine the biological behavior of VEGF-A. All isoforms contain a receptor binding site within amino acids 81 through 92, whereas only the longer isoforms (165 and longer) contain the heparin binding site within amino acids 110 through 165. The heparin binding site interacts with matrix and cell membrane proteoglycans, thereby causing longer isoforms to sequester, and allows diffusible VEGF165 to interact with neuropilins, membrane-bound coreceptors, which favorably present VEGF to the transmembrane receptors and amplify by 10-fold the resultant biological activity.
Several lines of evidence have linked VEGF to the development of ocular vascular conditions. Soon after the discovery of VEGF, investigators detected elevated intraocular concentrations in eyes suffering from several different chorioretinal vascular diseases.43,44 Not surprisingly, eyes with large areas of retinal ischemia (ie, those at highest risk of developing retinal and iris neovascularization) have the highest VEGF concentrations. VEGF-A, VEGF-B, and placental growth factor have all been detected in choroidal neovascular membranes.45–47 The administration of exogenous VEGF produces a clinical picture with retinal hemorrhages, vascular dilation, and neovascularization that is indistinguishable from diabetic retinopathy. 48 Matrigel, a VEGF-rich, gelatinous protein mixture secreted by Engelbreth-Holm-Swarm mouse sarcoma cells, reliably produces choroidal neovascular membranes in mouse models. In experimental models, selective inhibition of VEGF prevents the development of laser photocoagulation-induced choroidal neovascular membranes and fibroblastic growth factor–induced corneal neovascularization.
Anti-VEGF Drug Development
Each of the available anti-VEGF drugs works by binding diffusible VEGF dimers, thereby preventing their activation of the transmembrane VEGF receptors (Fig. 1). The first commercially available anti-VEGF drug was pegaptanib (Macugen®, Eyetech, Palm Beach Gardens, FL), a high binding affinity (KD = 50 pM for VEGF165) aptamer to VEGF165, which was pegylated (addition of a 40 kDa polyethylene glycol chain) to both prevent premature cleavage by endonucleases and to prolong its intraocular half-life. 49 Since pegaptanib interacts only with the heparin binding site of VEGF165, developers thought that it would optimally combine potent anti-VEGF activity with a low risk of unwanted anti-VEGF side effects. In phase 3 trials, pegaptanib reduced vision loss due to exudative AMD by one half but, unfortunately, few patients experienced improved vision. 50
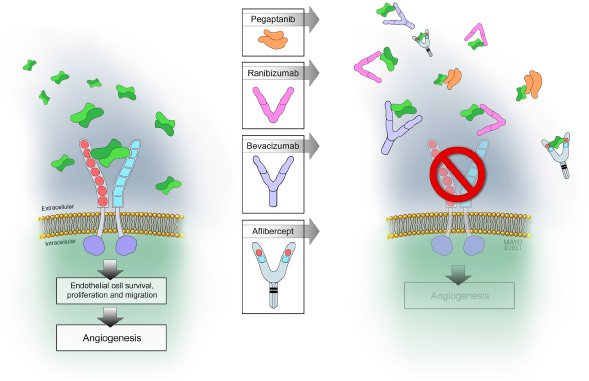
The left side of the figure shows diffusible VEGF dimers binding to and dimerizing VEGF receptors. (Originally published in CML-Ophthalmology 2012;22(4):106, reprinted courtesy of Remedica.)
Soon after pegaptanib was approved, bevacizumab (Avastin®, Genentech, S. San Francisco, CA/Roche, Basil, Switzerland), a full-length, humanized antibody to VEGF, was first injected into eyes with exudative AMD 51 and macular edema due to a central retinal vein occlusion. 52 The rapid resolution of edema and improvement in vision was so startling that off-label use of bevacizumab spread rapidly throughout the world and soon became the anti-VEGF drug most preferred by physicians.
Bevacizumab was developed for the treatment of advanced solid malignancies, but its developers recognized that VEGF suppression would also be valuable for chorioretinal vascular conditions. Because they were concerned that bevacizumab's large size (149 kDa) would prevent its diffusion from the vitreous through the inner retina and its Fc fragment would incite a destructive immunologic response and prolong the drug's systemic half-life, the VEGF binding sequences were isolated and affinity-enhanced to create ranibizumab (Lucentis®, Genentech, S. San Francisco, CA/Roche, Basel, SW), a humanized, VEGF binding fragment (Fab). 53 Phase 3 trials demonstrated that ranibizumab prevented vision loss better than observation in eyes with occult CNVM 54 and better than photodynamic therapy in eyes with predominantly classic membranes. 55 These impressive results convinced the US FDA to approve ranibizumab on June 30, 2006, for the treatment of all forms of foveal-involving exudative AMD.
This antibody-based technology created drugs with high binding affinities to all isomers of VEGF-A (bevacizumab: KD = 58 pM – 20 nM; ranibizumab: KD = 46 pM – 192 pM, for VEGF165), but scientists at Regeneron decided to use high affinity, native receptor binding sequences and recombinant technology to create a soluble decoy receptor. Since VEGFR1 possesses a higher affinity for VEGF than does VEGFR2, a “parent” trap (VEGF TrapR1R1RR1) consisting of the first 3 binding domains from VEGFR1 fused to an Fc fragment of IgG1 was created. As expected, this molecule bound VEGF effectively (KD = 5 pM), but, because of its basic protein sequences, it sequestered rapidly within the intercellular matrix and exhibited poor pharmacokinetic behavior. Therefore, subsequent traps (VEGF TrapδB1 and VEGF TrapδB2) were synthesized to decrease matrix binding and improve bioavailability. The inclusion of the third VEGF binding domain from VEGFR2 together with the second binding domain from VEGFR1 finally created a molecule (VEGF TrapR1R2, aflibercept) with minimal matrix binding and excellent pharmacokinetic behavior. 56 Furthermore, 3-dimensional stoichiometric modeling suggested that the molecule's two variable regions were able to tightly (KD = 0.5 pM) bind each of the VEGF dimer's constituent molecules in a “two-fisted” grasp with a strength that exceeded even that of the parent trap.
The results of biological activity assays comparing aflibercept with ranibizumab and bevacizumab have been mixed. In one study, aflibercept inhibited vascular endothelial cell proliferation and migration similarly to ranibizumab, and both were 11 times as effective as bevacizumab. 57 In another, aflibercept was 10 to 129 times as effective as bevacizumab and ranibizumab at preventing endothelial cell migration and calcium mobilization. 58 One investigator opined that these differences were due to the different VEGF concentrations used in the assays. 58
Once aflibercept's binding sequences had been set, drug development proceeded by targeting both oncologic and ophthalmologic conditions. Preclinical studies showed that aflibercept prevented leutinization in marmosets 59 and significantly decreased the growth of several orthotopic tumors in mice. 60 It prevented choroidal neovascularization in laser photocoagulation and matrigel models in mice, 61 decreased basic fibroblast growth factor–induced corneal neovascularization, 62 and increased the survival of high risk corneal transplants. 63
The intravenous administration of aflibercept results in rapid and nearly irreversible binding of VEGF with serum half-lives of 1 to 3 days for free aflibercept and 18 days for the bound complex. The Fc fragment prevents filtration by the kidneys, thereby requiring that the drug be removed by pinocytotic mechanisms. 64 The intravitreal half-life of aflibercept in rabbits is 4.7 days, 65 longer than that of both ranibizumab (2.8 days) and bevacizumab (4.2 days).66,67
Clinical Trials
Aflibercept's impressive performance in preclinical studies convinced investigators to proceed with human oncologic and ophthalmologic trials. The first ophthalmologic trial randomized 25 patients with subfoveal CNVM and visual acuity worse than 20/40 to receive 3 doses of intravenous aflibercept (0.3 mg/kg [7 patients], 1 mg/kg [7 patients], or 3 mg/kg [5 patients]) or placebo (6 patients). 68 After the first dose, patients were observed for 1 month to ensure safety and then were given 3 additional doses at 2 week intervals. The mean changes in excess retinal thickness for the placebo, 0.3 mg/kg, 1 mg/kg, and 3 mg/kg groups were, respectively, -12%, -10%, -66%, and -60%. A significant improvement in visual acuity was not seen in this small study. The most common side effects were hypertension, proteinuria, and headaches. Side effects in the groups receiving 1 mg/kg were easily managed, but hypertension and proteinuria in patients receiving 3 mg/kg were severe. One patient with severe hypertension responded slowly to changes in antihypertensive medications and developed congestive heart failure with pulmonary edema. Therefore, 1 mg/kg was determined to be the highest tolerated dose of aflibercept. Unbound aflibercept in the plasma was found after day 14 only in patients receiving 3 mg/kg, suggesting that doses of 1 mg/kg and lower are insufficient to bind VEGF through 1 month. Due to these safety concerns over high dose intravenous aflibercept, the only dose that was sufficient to suppress CNVM activity for 1 month, all subsequent ophthalmologic trials were performed with intravitreal injections.
The phase 1 Clinical Evaluation of Anti-Angiogenesis in the Retina Intravitreous Trial (CLEAR-IT 1) evaluated the safety, tolerability, maximum tolerated dose, and bioavailability of intravitreal aflibercept. 69 Twenty-one patients with subfoveal CNVM under 12 disc areas in size and visual acuity less than 20/40 received single injections of the following 6 doses of aflibercept: 0.05 mg (3 patients), 0.15 mg (3 patients), 0.5 mg (3 patients), 1 mg (6 patients), 2 mg (3 patients), and 4 mg (3 patients). Intraocular injections began with the lowest dose and only after a 1-month observation period established tolerance was the next dose administered. All doses were well tolerated with no evidence of intraocular inflammation or toxicity, so the maximum tolerated intravitreal dose was not determined. The primary endpoint was set at 6 weeks but patients were followed for 12 weeks.
Average visual acuities improved by +4.43 letters at 6 weeks, but for the two groups receiving the highest doses (2 mg and 4 mg), the average gain was +13.5 letters. Three of these 6 patients improved by >15 letters, and 3 required no additional treatment through 12 weeks. The mean decrease in foveal thickness at 6 weeks was -104.5 μm for all groups, whereas it was substantially more (-216.1 μm) for the groups receiving the 2 highest doses (2 mg and 4 mg). The CLEAR-IT 1 trial showed that most patients receiving intravitreal aflibercept responded well for at least 6 weeks and that patients receiving the highest doses exhibited the greatest improvements in vision and thinning.
The phase 2 CLEAR-IT 2 trial evaluated the biological effects and safety of aflibercept during an initial 12-week fixed-dosing period followed by a 40-week period with PRN dosing.70,71 One hundred fifty-nine patients were randomized 1:1:1:1:1 to receive monthly injections (0.5 mg or 2 mg) or quarterly injections (0.5 mg, 2 mg, or 4 mg) for the first 12 weeks. Enrolled patients had subfoveal CNVM < 5400 μm in diameter, central retinal lesion thicknesses > 300 μm, and visual acuities from 20/40 to 20/200. Both the primary (change in central retinal lesion thickness) and secondary (change in best corrected visual acuity) endpoints were measured at week 12, but additional measurements were made at week 16 to assess the effects of the final fixed dose administered at week 12. Additional endpoints included avoidance of significant loss in vision (loss of <15 letters), stabilization of vision (change of >0 letters), and significant improvement in vision (gain of > 15 letters).
Patients improved quickly after the first injections as the average gains in vision (+3 letters) and excess macular thickness (-103 μm, P = 0.04) were significant at 1 week. The average gain in vision at week 12 was +5.7 letters but patients receiving monthly injections gained more (0.5 mg: +8.8 letters; 2 mg: +8.3 letters). Ninety-eight percent of patients avoided significant vision loss (<15 letters) and 19% achieved significant visual gains (26% of those receiving 2 mg every 4 weeks). Not surprisingly, the proportion of patients seeing worse than 20/200 decreased, and the proportion seeing better than 20/40 increased, with the greatest proportion (58%) among those receiving 2 mg every 4 weeks. At 8 weeks, the average gains in vision were similar between patients receiving 2 mg every 4 weeks and 2 mg every 12 weeks, but greater thinning was seen in the monthly group. The average decrease in central retinal/lesion thickness at 12 weeks was -119 μm, but eyes receiving monthly injections had significantly more thinning (0.5 mg every 4 weeks: -153.5 μm; 2 mg every 4 weeks: -169.2 μm) than those receiving quarterly injections.
After the mandatory injection at week 12, all treatment groups continued to improve through week 16. Improvements in average visual acuity (VA) (+6.6 letters), macular thinning (-160 μm), and the proportion of patients gaining > 15 letters (23% of all patients; 39% in the 2 mg every 4 weeks group) were noted.
Most ocular adverse events were related to the injections, and none were severe. Twelve patients experienced systemic adverse events including congestive heart failure and coronary artery disease, but none were believed to be drug-related.
During the second phase of the CLEAR-IT 2, patients were examined monthly and treated if neovascular activity in the form of edema or subretinal fluid, new hemorrhage, leaking on fluorescein angiography, or a decrease of more than 5 letters of vision was discovered. The most common reason for reinjection was persistent fluid (63%) on optical coherence tomography (OCT). By week 52, patients improved an average of +5.3 letters from baseline with the greatest gains (+9 letters) enjoyed by patients who originally received 2 mg every 4 weeks. The average improvement in macular thickness was -130 μm, whereas the group that initially received 2 mg every 4 weeks improved by -143 μm. During phase 2, patients required an average of 2 additional aflibercept injections, with the first being given an average of 129 days after the completion of phase 1. During the PRN phase, 19% of patients required no additional injections, and 45% needed only 1 or 2 injections. By the end of the trial, the area covered by the neovascular membranes regressed by an average of 2.21 mm3 (approximately 30%). Moderate vision loss (<15 letters) was avoided by 92% of the study patients (range, 88%–100%), stabilization of vision (change of >0 letters) occurred in 73.5% of patients, and 22% (29% of patients receiving 2 mg) were significant gainers (> 15 letters). Forty-one percent of patients achieved a final vision of at least 20/40. Most ocular adverse events were mild with conjunctival hemorrhage being the most common (38%). Thirty-five patients experienced 57 severe adverse events, but none were considered to have been due to the study drug. Four ocular severe adverse events were reported (study eye: culture negative endophthalmitis and loss of vision; fellow eye: retinal detachment and increased intraocular pressure), and 1 patient suffered a cerebral vascular accident.
The CLEAR-IT 2 trial demonstrated that the higher doses of aflibercept (2 mg and 4 mg) produced superior improvements in vision and thinning compared to lower doses. After a 12-week fixed dosing period, improvements in vision and edema could be maintained by PRN dosing, though few injections (average of 2) were required during the subsequent 40 weeks. The trial results suggested that although a monthly loading regimen produced similar results to single dosing at 8 weeks, it produced better long-term results. These lessons were used to design the phase 3 trials.
The two parallel phase 3 VEGF Trap-eye: Investigation of Efficacy and Safety in Wet AMD (VIEW 1 and VIEW 2) trials evaluated the efficacy and safety of different doses (0.5 mg and 2 mg) and treatment regimens (every 4 weeks and every 8 weeks after 3 monthly injections) of intravitreal aflibercept compared with monthly ranibizumab. 72 Previously untreated patients with subfoveal CNVM due to exudative AMD and visual acuities from 20/40 to 20/320 were randomized 1:1:1:1 to receive 1 of 3 doses of aflibercept (0.5 mg every 4 weeks, 2 mg every 4 weeks, or 2 mg every 8 weeks) or ranibizumab every 4 weeks. All patients were examined and treated monthly, whereas patients receiving aflibercept every 8 weeks were treated with 3 consecutively monthly injections followed by sham injections every other visit (Fig. 2). A total of 2457 patients were randomized–-2419 completed the 52-week trial–-at sites in North and South America, Europe, Asia, and Australia. The primary endpoint was noninferiority (defined as less than a 10% difference) of aflibercept compared with ranibizumab for the prevention of moderate vision loss (loss of <15 letters). Important secondary endpoints included mean change in best corrected visual acuity, proportion of patients gaining at least 15 letters, improvement on the National Eye Institute (NEI) VQF-25 questionnaire, which assesses vision-related quality of life, and change in the fluorescein angiographic-defined area of the CNVM.
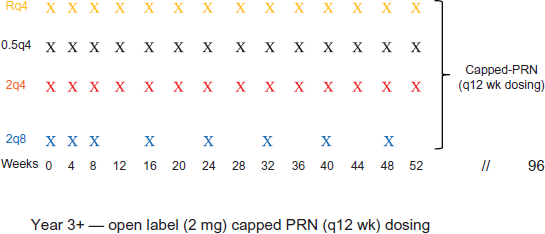
This figure shows the treatment schedule for the 2 years of the VIEW studies as well as the extension study which began in year 3.
The trials’ primary endpoint was easily met as 95% to 96% of patients receiving aflibercept and 94% of those receiving ranibizumab avoided moderate vision loss. Integrated data from both trials showed that the improvements in best corrected visual acuity were similar for patients receiving aflibercept and those receiving ranibizumab (+8.3 to +9.4 letters vs. +8.7 letters) (Table 1). All aflibercept groups from each trial achieved comparable gains in vision compared to those treated with ranibizumab (+6.9 to +10.9 letters vs. +8.1 and +9.4 letters) except for the group receiving 2 mg every 4 weeks in the VIEW 1 (+10.9 vs. +8.1 letters; P < 0.005). Since the 2 mg every 4 weeks group improved the least in the VIEW 2 (+7.6 letters), the VIEW 1 differences were felt to be a statistical aberration. Similar proportions of patients receiving aflibercept and ranibizumab gained at least 15 letters (24.9% to 37.5% vs. 30.9% and 34%). Also, the magnitude of central retinal thinning was similar among patients receiving aflibercept (-115.6 μm to -156.8 μm) and ranibizumab (-116.8 μm and -138.5 μm). Sawtooth variations in retinal thickness, beginning at 17 μm and decreasing to 8 μm by the completion of the VIEW 2 trial, were seen every other month, but coincident changes in vision did not occur. Dry retinas were achieved in most patients receiving aflibercept (2 mg every 4 weeks: 72.4%; 0.5 mg every 4 weeks: 60.3%; 2 mg every 8 weeks: 67.7%) and ranibizumab (62.0%). Scores on the NEI VFQ-25 were similar among all groups.
Selected results from years 1 and 2 of the VIEW trials are tabulated.

Aflibercept exhibited an excellent safety profile with few serious ocular events (endophthalmitis, increased intraocular pressure, and procedural complications) seen in the groups receiving 2 mg every 4 weeks (0.8%), 0.5 mg every 4 weeks (0.1%) and 2 mg every 8 weeks (0.2%) when compared with those receiving ranibizumab (1.1%).
The incidence of ocular and nonocular treatment emergent adverse events (TEAEs) were similar among all study groups and were nonsignificantly lower in patients treated with aflibercept in the VIEW 1 but were slightly higher in aflibercept-treated patients in the VIEW 2. There were 3 patient deaths, 1 from each of the aflibercept groups. The integrated data showed similar rates of TEAEs in all groups with the lowest rates among patients receiving aflibercept 2 mg every 4 weeks. Cardiac disorders occurred with equal and low frequencies among all treatment groups (ranibizumab: 3.4%; aflibercept: 3.2%). The incidence of systemic arterial hypertension (0.3%), often felt to be the most sensitive sign of systemic VEGF suppression, was the same in patients receiving ranibizumab and aflibercept.
Nervous system disorders were seen in 1.7% of all patients, with transient ischemic attacks (TIAs) (0.4%) and cerebrovascular accidents (CVAs) (0.3%) occurring most commonly. The incidence of nervous system disorders was lower in the ranibizumab group (0.5%) than in the aflibercept groups (1.5% to 2.5%), but none of the patients with the greatest drug exposure (aflibercept 2 every 4 weeks) experienced (TIAs).
The analysis plan specified that a broad range of terms, “soft” events including TIAs and “hard” events including CVAs and myocardial infarctions, would be included in the definition of potential arterial thromboembolic events. The original analysis calculated an incidence of events that ranged from 1.3% in the ranibizumab group to 2.5% in the pooled aflibercept groups. A second analysis of the ATEs was performed by a masked panel of experts who applied criteria approved by the Antiplatelet Trialists’ Collaboration. According to these criteria, ATEs were defined as myocardial infarction, nonfatal ischemic stroke, nonfatal hemorrhagic stroke, or death due to vascular or unknown causes. This analysis showed no significant difference between ATEs due to ranibizumab (1.5%) and aflibercept (1.8%) with the highest dose group (aflibercept 2 mg every 4 weeks) having the lowest rate (1.0%). They concluded that no significant differences in ATE rates existed between aflibercept and ranibizumab treated patients. 73
Patients were examined monthly during the second year of the VIEW trials and treated with the same drug and dose as during the first year when signs of neovascular activity (persistent or recurrent edema or subretinal fluid, increased macular thickness of >100 μm, decreased vision of >5 letters, evidence of active neovascularization on fluorescein angiography) were discovered. However, investigators were concerned that long periods without VEGF suppression might compromise visual outcomes so a 12-week cap was placed on the treatment interval. Therefore, between weeks 52 and 96, patients were required to receive a minimum of 3 injections. Patients originally randomized to aflibercept 2 mg every 8 weeks received an average of 4.2 injections, whereas patients receiving ranibizumab required 4.7 injections. Compared with those receiving ranibizumab, a smaller proportion of patients receiving aflibercept required the most intensive treatment (11 or more injections: 3.0% vs. 1.8%; 6 or more injections: 25.6% vs. 15.9%). Patients originally receiving both aflibercept 2 mg every 8 weeks and ranibizumab lost an average of only 0.8 letters of best corrected visual acuity (+8.4 to +7.6 vs. +8.7 to +7.9) during the second year. 74
The first 2 years of the VIEW trials demonstrated that aflibercept has a longer duration of action than ranibizumab and, by extension, bevacizumab. Unfortunately the 12-week capped PRN protocol during year 2 prevented an accurate determination of aflibercept's average duration of action. Since 48% of patients required only the minimum number of injections, the average (median) duration of action appears to be around 12 weeks. This is consistent with mathematical modeling that predicted an average duration of action of between 2.5 and 3 months (Fig. 3). 75 This longer duration of action should translate into fewer patient visits to the physician's office.
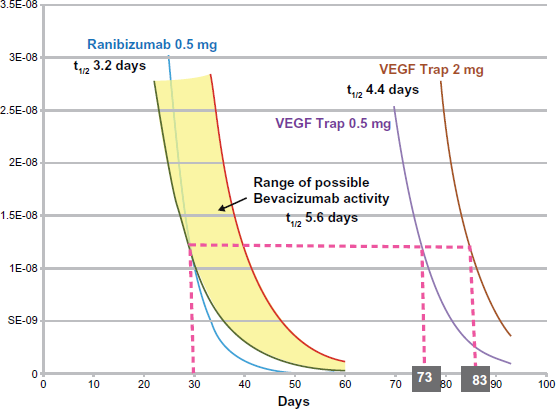
Mathematical modeling suggests that the VEGF binding capacity of intraocular aflibercept (VEGF-Trap) has a duration of action of just under 3 months, longer than both ranibizumab and bevacizumab.
Since aflibercept received regulatory approval, its use by physicians has exceeded the expectations of most analysts. The American Society of Retina Specialists surveyed its members regarding anti-VEGF drug use and published its findings in the 2012 Preferences and Trends manuscript. A total of 631 surgeons responded to the survey, with approximately 15% of them from outside of North America. Aflibercept had not been used by 22.6% of respondents, and only 10.8% viewed aflibercept as the drug of first choice for exudative AMD, but 63.6% of specialists were trying it on select patients with persistent fluid, edema, or RPE detachments. Although 66.5% of physicians prefer bevacizumab as their first choice drug, 55.7% would choose aflibercept if the available AMD drugs cost the same.
At the time of this writing, no postapproval aflibercept manuscripts with prospectively acquired data have been published so we are unable to independently confirm aflibercept's duration of action. In addition to its use as a first-line therapy, aflibercept has emerged as an effective “salvage” therapy for eyes that have responded incompletely to bevacizumab and ranibizumab. At the 2012 American Society of Retinal Specialists meeting (Las Vegas, NV, August 25–29, 2012), 6 speakers, Vincent Hau, Patrick Williams, Ashish Sharma, Harold Wheatley, Irene Barbazetto, Kirk Packo, discussed aflibercept use as salvage therapy. Eyes that were previously treated with bevacizumab or ranibizumab experienced improved vision (20/68 to 20/61) and macular thinning (336 μm to 275 μm) when given only 1 or 2 injections of aflibercept.
The reason for aflibercept's effectiveness in eyes that have incompletely responded to previous anti-VEGF therapy is unknown but several theories have been advanced. Since aflibercept has a higher affinity for VEGF, it leaves a lower unbound VEGF concentration. In addition to binding all isoforms of VEGF-A, aflibercept binds VEGF-B and placental growth factor, both of which have been identified in CNVM. Finally, it may be that simply changing medications and avoiding possible adverse immunological reactions may be beneficial.
After the first 30,000 postapproval aflibercept injections, 14 cases (11 from the same practice and 9 from a single physician) of sterile intraocular inflammation had been reported to Regeneron. An investigation into the manufacturing, packaging, shipping, and storage of the drug, as well as the administration technique used by the physicians, failed to uncover the cause of the inflammation. Inflammation occurred with doses from different manufacturing lot numbers, and other physicians reported no inflammation when using doses from the same drug lots. This series of cases prompted a “Dear doctor” letter to inform retinal physicians of these problems. 76 Since these early reports, no subsequent clusters of inflammation have been reported, and the overall incidence of posta-flibercept inflammation has been comparable to that seen with other intravitreal drugs.
In the United States, aflibercept's wholesale price of $1850 per dose is slightly less than that of ranibizumab ($1950) but still far greater than that of bevacizumab (approximately $50–$80 per dose). Costs for the first year of protocol-driven anti-VEGF therapy vary considerably by drug: the cost of ranibizumab is $29,582, the cost of bevacizumab, $4745, and the cost of aflibercept, $18,566. However, costs incurred with PRN and treat-and-extend protocols are much lower and are less dependent upon the choice of drug. The costs for ranibizumab, bevacizumab, or aflibercept therapy are as follows, respectively: PRN would be $10 832, $2042, and $9273 and treat-and-extend would be $11 961, $2236, and $8842.
In addition to the United States, aflibercept has received regulatory approval for the treatment of exudative AMD in Australia, Japan, and Europe. Based on excellent results from the COPERNICUS and GALLILEO trials, aflibercept was also approved in the United States for the treatment of macular edema due to central retinal vein occlusions. Phase 3 trials for the treatment of macular edema due to branch retinal vein occlusions and diabetic macular edema are ongoing.
Conclusion
Aflibercept has become a welcome addition to the expanding formulary of anti-VEGF drugs as it functions well as both first-line therapy and salvage treatment. Since we will probably need to wait 5 years until another anti-VEGF drug becomes available, aflibercept provides physicians and patients a valuable alternative to existing therapy.
Author Contributions
Conceived and designed the experiments: MWS. Analyzed the data: MWS. Wrote the first draft of the manuscript: MWS. Contributed to the writing of the manuscript: MWS. Agree with manuscript results and conclusions: MWS. Jointly developed the structure and arguments for the paper: MWS. Made critical revisions and approved final version: MWS. All authors reviewed and approved of the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
MWS has received consulting fees from Boehringer-Ingelheim, is an advisory board member for Allergan, and is an advisory board member and has received research support from Regeneron.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Provenance: the authors were invited to submit this paper.