
Abstract
The burden of disease related to chronic hepatitis C (CHC) continues to increase annually. While our experience with treating CHC began less than 30 years, steady progress has been made in the ability to successfully treat patients, reducing morbidity and mortality. Until recently, the main players in therapy of CHC were interferon and ribavirin. Unfortunately, response to this therapy is successful only in a selected group of patients, leaving a sizeable portion of patients with CHC untreated or with ineffective retreatment options after having failed prior therapy. An in depth understanding of the hepatitis C virus has ushered in the dawn of a new era of therapy for CHC. Two drugs, telaprevir and boceprevir, have recently been approved by the Food and Drug Administration. Many others hold great promise and are in the early phases of drug development. Here, we will review the history of hepatitis C therapy, mechanism of action drugs approved for or in development, current data on clinical safety and efficacy of these agents as well as the role of patient preference in CHC therapy. We will conclude with current recommendations for the treatment of patients with CHC and the evolving role of interferon and ribavirin.
Keywords
Introduction
In recent years, deaths associated with the hepatitis C virus (HCV) have overtaken deaths caused by HIV. 1 It is estimated that 3% of the world's population (130-170 million people) are chronically infected with HCV. 2 The most recent National Health and Nutrition Examination Survey (NHANES) found that 1.3% of the US population, or more than 3 million people, have chronic hepatitis C (CHC). 3 Prevalence of CHC peaked in 2001 at 3.6 million, while the prevalence of HCV related cirrhosis and its complications are on the rise and reported to peak in 2020 at 1 million people in the US. 4 Initiation of treatment is aimed at reducing the high morbidity and mortality associated with the natural history of chronic infection, particularly, the development of hepatic fibro-sis, cirrhosis, and their ensuing complications such as portal hypertension and hepatocellular carcinoma.
The history of hepatitis C therapy
Therapy for chronic hepatitis C infection has been an evolving process since the first published investigation of interferon-alfa (IFN) therapy in 1983. 5 At that time treatment success for non-A, non-B hepatitis was determined by trending transaminase values. The year 1989 marked a milestone in therapy, when the first prospective randomized control trial assessing the efficacy of interferon-alfa revealed a 24-week course of tri-weekly IFN gave histologic and serologic improvement at 6 months in 40%-50% of those treated, compared to 8% of those untreated.6,7 The same year Houghton et al identified an assay that could be used to detect HCV ribonucleic acid (RNA) and allow measurement of virologic response to treatment.8,9
Four types of interferon were under development in the early 1990's: recombinant interferon- alfa-2b (IFN-2b) and interferon-alfa-2a (IFN-2a), a monoclonal antibody-purified lymphoblastoid alpha interferon named interferon lymphoblastoid, as well as a beta interferon. 9 In 1991, IFN was approved for Hepatitis C therapy. Studies using the new HCV RNA assay showed low (<20%) overall rates of sustained virologic response (SVR) with IFN monotherapy. 7 Attempts to prolong the treatment course to 1 or 2 years were made with minimal gain in SVR (35%), and with great expense in the form of adverse effects, cost, and inconvenience of treatment. 9 In 1992, several investigators from Japan identified different strains of HCV and were first to report significantly different responses rates to IFN among genotype populations.10–12
Ribavirin (RBV) is nucleoside analogue with known activity against several flaviviruses. When used alone, RBV improved transaminase levels and histologic response, but had minimal effect on HCV RNA levels. Two landmark studies comparing IFN-2b and IFN-2a given in combination with RBV for 48 weeks produced SVR rates of 40%-50%, 2 to 3 times those obtained with interferon alone. The SVR rate ranged from 16%-28% in patients with genotype 1, and 66%-69% in those with genotypes 2 or 3.13,14
A third advancement in hepatitis C therapy came soon after, with the production of a covalent attachment of polyethylene glycol to the interferon molecule. With its increased half-life, pegylated interferon-alfa (PEG-IFN) could be given as a weekly dose. In two large trials of these agents, the rates of SVR to a 48-week course of PEG-IFN and RBV were 54% and 56%, as compared with 44% and 47% with standard interferon and RBV, and only 29% with PEG-IFN alone. Again, response rates were higher among patients with genotype 2 or 3 than among those with genotype 1. A subsequent trial of different regimens of PEG-IFN-2a and RBV showed that patients with genotype 2 or 3 could be treated with a lower dose of RBV (800 mg rather than 1000-1200 mg daily), and that SVR rates after 24 weeks of therapy (81% and 84%) were similar to the rates after 48 weeks of therapy (79% and 80%).8,15–18
With the recent approval the first of several novel agents coined direct acting antivirals (DAAs), treatment has now evolved beyond stimulation of the host immune system and non-specific targeting of viral replication. Greater understanding of the HCV genome and life cycle of the HCV viron allows for new targets for therapy that directly act on the viral machinery to inhibit replication. (Fig. 2) There are currently two protease inhibitors that are approval by the Food and Drug Administration (FDA) for standard use in combination with PEG-IFN and RBV, boceprevir and telaprevir, both of which will be discussed at length below. In addition, many more DAAs are in development, many soon to be approved for use to treat chronic hepatitis C with and without PEG-IFN and RBV. With the improved efficacy afforded by the DAAs, triple therapy (PEG-IFN/RBV plus a protease inhibitor) has become the new standard of care for genotype 1 HCV
Responses to therapy and terminology
While patient characteristics such as viral genotype, viral load, IL28B genotype and race are important at predicting treatment outcome, a patient's viral kinetics in response to treatment is the most accurate predictor of treatment outcome. The established conventions describing treatment response were created to ensure universality in studying treatment efficacy with PEG-IFN/RBV. While some of the following viral responses will lose importance to newly defined viral kinectics in the era of DAAs, it is important to understand the old definitions as they continue to serve as the backbone of defining treatment response for most patients.
Responses are defined according to their timing relative to the course of treatment (Fig. 1). SVR defined as the absence of HCV RNA from serum 24 weeks following discontinuation of therapy, is generally regarded as “virologic cure,” as relapse occur in only 1%-3% of patients. 19 A rapid virologic response (RVR), or HCV RNA negative at week 4 of treatment, predicts high likelihood of SVR (85%-90%). 20 Conversely, failure to achieve an early virologic response (EVR), or >2 log reduction in HCV RNA at treatment week 12, predicts failure to achieve SVR in 97%-100% of patients.21,22 Partial EVR (pEVR) is defined as a >2 log drop of HCV RNA at treatment week 12 compared with baseline, while complete EVR (cEVR) describes unde-tectable HCV RNA at treatment week 12 compared with baseline. For patients treated with triple therapy, extended RVR (eRVR), defined as undetectable HCV RNA both at week 4 and week 12, was also found to be highly predictive of SVR. In addition, new data suggests that patients achieving eRVR, triple therapy can shorten therapy to 24 weeks without decline in SVR rates.
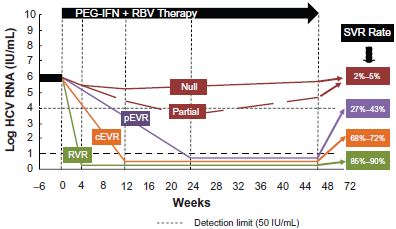
Viral kinetics on therapy are the best predictors of viral eradication. Sustained virologic response (SVR) is considered virological cure. Rapid virologic response (RVR) is achieved in about 20% of patients and helps predicts SVR. Early virologic response (EVR) is achieved in about 50% of patients but failure to do so is a strong marker for treatment failure. Patients experiencing nonresponse make up a third of treated patients and are separatedinto partial responders and null nonreponsders. Both groups are very unlikely to achieve SVR, however, partial responders fair better when retreatment is attempted.

HCV life cycle and multiple therapeutic targets. Host proteases release the structural HCV protein core (C) and envelope 1 and 2 (E1, E2) proteins. A combination of host and viral proteases process 6 nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B), whose enzymatic functions are obligatory for the HCV life cycle. NS2 and NS3 are viral proteases required for HCV polyprotein processing. The NS3/NS4A protease complex is required for proteolysis of the polyprotein to release NS5A and NS5B. The replication complex required for HCV RNA synthesis is shown in greater detail on the right. NS5B is the RNA-dependent RNA polymerase responsible for the synthesis of a complementary negative strand RNA from the positive-strand RNA genome. From the complementary negative-strand RNA, multiple positive-strand RNA molecules are produced and used as messenger RNAs for production of polyprotein, as new templates for negative strand synthesis, and as genomic RNA in newly formed virions. Because of the high error rates in RNA replication, numerous HCV quasispecies, are produced.
Nonresponders include null responders, partial responders, and those who experience breakthrough during treatment. Null responders fail to decrease their HCV RNA by 2 logs after 24 weeks of therapy, or have < 1 log decline in HCV RNA at 4 weeks of therapy (early null responder). Early null response is associated with 92% negative predictive value for SVR. 23 Partial responders decrease their HCV RNA by 2 logs from their baseline but remain HCV RNA–-positive at week 24 of therapy. Patients who experience breakthrough are those who initially cleared virus with therapy but have replication resume despite continued treatment. A relapser is defined as a patient who experiences a reappearance of HCV RNA in serum after therapy is discontinued despite having cleared the virus at the end of treatment. 3
Factors predicting treatment response
The decision to start treatment requires a balance between efficacy, need and safety. Each patient has a unique risk to benefit ratio when considering therapy for CHC and it is difficult to asses which patients with CHC will progress to more significant liver disease. Therefore, many factors must be considered in whether or not to pursue or re-pursue treatment.
Viral factors are considered first, the most important of which is genotype, as this is the strongest baseline predictor of response. As discussed above, patients with genotypes 2 and 3 have SVR rates of 70%-80% with 24-week standard PEG-IFN/RBV, while the genotype 1 population achieves SVR 40%-50% of the time with a 48-week treatment course. Baseline HCV load is also an important predictor of response, as lower viral loads (<400,000 IU/mL to 800,000 IU/mL) respond more frequently. Host factors are of great importance as well. Poor treatment response has been associated with male gender, high body-mass index, hepatic steatosis, and insulin resistance. Ethnicity is also an important consideration. Observations that African Americans respond half as frequently as Caucasians, whileAsians respond more frequently than Caucasians, led to the discovery that a genetic polymorphism (rs12979860) near the Interleukin (IL)-28B (IL28B) gene portends a poorer response to therapy. 24 Finally, it remains that patients with advanced fibrosis not only gain the most benefit from SVR, they are also the most challenging to treat as they tolerate therapy more poorly and continue to have suboptimal SVR rates. It is important to note that as therapeutic options for CHC are evolving with the development of DAAs, these factors are likely to carry less weight in both the decisions to treat CHC and their impact on treatment response.
Co-infection with human immunodeficiency virus (HIV) is another important consideration, as rates of progression to cirrhosis, end-stage liver disease, and hepatocellular carcinoma are all increased in coinfected individuals. Mortality rates are 35% greater in HCV/HIV infected patients than HIV infection alone, regardless of HIV severity. The importance of CD4 count influencing SVR rates has yielded inconsistent results. Nonetheless, consensus agrees attempts to increase CD4 counts prior to initiation of HCV therapy should be pursued, but therapy should not be deferred if unsuccessful. 25
Genetic variation of the IL28B gene on chromosome 19, encoding interferon-lambda-3, has been shown to strongly correlate with outcomes in those with genotype 1 treated with PEG-IFN/RBV. Patients who have the CC genotype at rs12979860 have a twofold higher likelihood of experiencing an SVR compared with patients with either the CT or TT haplotype, regardless of race or ethnicity. In addition, favorable allele combinations were associated with spontaneous clearance of HCV infection, and in genotypes 2 and 3, the CC allele is more predictive of SVR overall. 26 Importantly, favorable IL-28B alleles (CC) are seen in greater frequency in Caucasians than in African Americans (39% vs. 16%). There is currently evolving work studying the interaction with IL-28B and the DAA's. Many clinics routinely incorporate IL-28B testing into treatment heuristics, which is available as a licensed diagnostic tool in North America (Labcorp, Burlington, NC, USA). 25
Standard of care
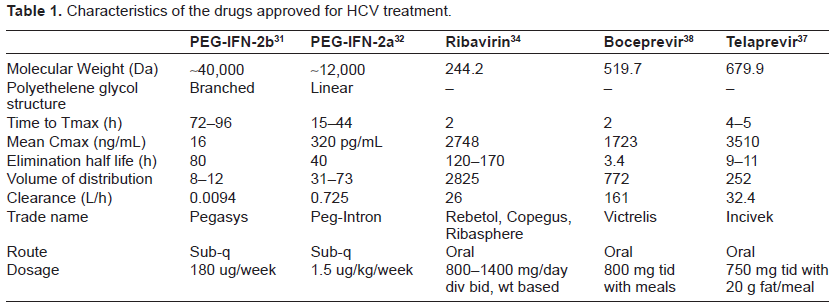
Prior to May 2011, the standard of care was combination PEG-IFN and RBV. Two PEG-IFN formulations are currently approved for the treatment of hepatitis C (Table 1): PEG-IFN-2a (Pegasys, Roche) and PEG-INF-2b (Peg-Intron, Schering-Plough). The recommended dose of PEG-IFN-2b is 1.5 µg/kg ideal body weight per week 16 and that of PEG-IFN-2a is 180 µg per week. 18 The optimal duration of therapy and the RBV dose vary according to genotype. Those with genotype 1 should receive 48 weeks of RBV at a dose of 1000 mg/day or 1200 mg/day if their weight is greater than 75 kg.16–18 The same treatment regimen is recommended for patients with genotype 4, 5 or 6 given the paucity of therapy data. Patients with genotype 2 or 3 infection should receive 24 weeks of combination therapy with a RBV dose of 800 mg daily.27,28 The majority of head-to-head randomized controlled trials, including the large, randomized IDEAL trial, have demonstrated similar SVR rates for PEG-IFN-2a and PEG-INF-2b in combination with RBV (41% vs. 39%, respectively). 8
Characteristics of the drugs approved for HCV treatment.
Mechanisms of Action, Metabolism and Pharmacokinetic Profiles
Pegylated interferon-alfa
Interferons are glycoprotein cytokines, which are named after their ability to “interfere” with viral replication within host cells. Interferon alfa and beta bind to cell membrane receptors and activate STAT (signal transducer and activator pathway transcription) complexes, the most well-defined of which is known as the Janus kinase-STAT (JAK-STAT). This signal cascade promotes nuclear transcription, which has three major downstream effects. First, resistance to viral replication is induced by activating cellular genes that destroy viral mRNA and inhibit the translation of viral proteins. Second, MHC class I expression is stimulated, thus increasing antigen presentation to CD8 cytotoxic T cells, which subsequently target cells harboring virus for cell death. Lastly, IFN induces cellular changes that make the virus infected cell more likely to be attacked by NK cells.20,29
The branched, 40-kDa polyethylene glycol chain of PEG-IFN-2a is covalently attached via stable amide bonds to lysine residues of PEG-IFN-2a, and circulates as an intact molecule. Consequently, PEG-IFN-2a has a longer half-life and reduced clearance compared with native IFN-2a, and can be given once weekly regardless of body weight. PEG-IFN-2b has a linear 12-kDa polyethylene glycol chain covalently attached primarily to histidine-34 of IFN-2b, releasing native IFN-2b. 30 PEG-IFN-2b has a shorter half-life in serum than PEG-IFN-2a and requires dosing based on body weight (Table 1).
Maximal serum concentrations (Cmax) for PEG-IFN-2a (Pegasys) occur between 72 to 96 hours post dose, and are sustained for up to 168 hours. The Cmax and AUC measurements increase in a dose-related manner (Table 1). Week 48 mean trough concentrations (16 ng/mL) are approximately 2-fold higher than week 1 mean trough concentrations (8 ng/mL). Steady-state serum levels are reached within 5 to 8 weeks of once weekly dosing. The mean systemic clearance in healthy subjects given was 94 mL/h, which is approximately 100-fold lower than that for non-pegylated IFN-2a. The mean terminal half-life after SC dosing in patients with chronic hepatitis C was 80 hours compared to 5.1 hours for the non-pegylated formulation. A 25%-45% higher exposure to peginterferon-α2a is seen in subjects undergoing hemodialysis. 31
With regard to PEG-IFN-2b (Peg-Intron), Cmax occurs between 15 to 44 hours post-dose, and are sustained for up to 48-72 hours. The Cmax and AUC measurements increase in a dose-related manner. Week 48 mean trough concentrations (320 pg/mL) are approximately 3-fold higher than Week 4 mean trough concentrations (94 pg/mL). Renal elimination accounts for 30% of the clearance. Single dose PEG-IFN-2b pharmacokinetics following a subcutaneous 1.0 µg/kg dose suggest the clearance of PEG-IFN-2b is reduced by approximately half in patients with impaired renal function (creatinine clearance <50 mL/min). 32
Treatment with PEG-IFN-2a is associated with a moderate inhibition of P450 1A2. There was no effect on the pharmacokinetics of representative drugs metabolized by CYP 2C9, 2C19, 2D6 or 3A4. 31 It is not known if PEG-IFN-2b therapy causes clinically significant drug-drug interactions with drugs metabolized by the liver in patients with hepatitis C. In 12 healthy subjects known to be CYP 2D6 extensive metabolizers, a single subcutaneous dose of 1 µg/kg PEG-IFN-2b did not inhibit CYP 1A2, 2C8/9, 2D6, hepatic 3A4 or N-acetyltransferase; the effects of PEG-IFN-2b on CYP 2C19 were not assessed. 32
Ribavirin
Ribavirin (RBV) is a pro-drug metabolized to resemble a purine RNA nucleotide, which subsequently interferes with viral replication. Its exact mechanism is not entirely clear. RNA viruses, such as hepatitis C, are thought to exist in populations that contain an ensemble of related genotypes, as opposed to a single “wild-type” virus. These “quasispecies” are thought to exist in a state of maximum variability, beyond which, the viral genetic information loses its meaning. The high genetic variability, which is normally a major advantage for an RNA virus, can be exploited by increasing the mutation rate beyond tolerable levels and causing a genetic meltdown. It has been proposed that incorporation of RBV into RNA need only induce moderate further mutation, leading to a so-called “lethal mutagenesis.” 33 Other actions have been proposed including weak polymerase inhibitor activity. Irrespective of mechanism, studies have repeatedly demonstrated the importance of RBV as dose reduction or disruption compromises SVR rates.
Multiple dose RBV pharmacokinetic data are available for HCV patients who received RBV in combination with PEG-IFN-2a (Table 1). Following administration of 1200 mg/day with food for 12 weeks mean Cmax was 2748 ng/mL. The average time to reach Cmax was 2 hours. The terminal half-life of RBV following administration of a single oral dose of RBV tablets is about 120 to 170 hours. There is extensive accumulation of RBV attributed to high intracellular erythrocyte concentrations such that the Cmax at steady state was four-fold higher than that of a single dose. The contribution of renal and hepatic pathways to RBV elimination after administration of RBV is not known. In vitro studies indicate that RBV is not a substrate of CYP450 enzymes and there is no evidence from toxicity studies that RBV induces liver enzymes. Therefore, there is a minimal potential for P450 enzyme-based interactions. 34
Boceprevir and telaprevir
Among the proteins that are encoded in the HCV RNA, there are structural and non-structural (NS) proteins. There are two structural proteins that have been identified, E1 and E2, which are necessary for fusion and entry into a cell membrane. Among NS proteins, the serine-like protease encoded in the NS3 region and the RNA-dependent RNA polymerase encoded in the NS5 region are among the many new targets for DAA compounds.35,36
A NS3 serine protease and a cofactor NS4A allow for post-translational cleavage of the 3000 amino acid polyprotein produced from HCV RNA and host ribosomes. Oncecleaved, this polyprotein forms 4 structural and 6 NS proteins key in viral RNA replication and new viral particle assembly. The NS3/4A complex is the direct target for both boceprevir and telaprevir. 37 Boceprevir (Victrelis™, Merck, Whitehouse Station, NJ, USA) is a novel peptidomimetic agent with a ketoamide structure that forms a covalent and reversible bond to the NS3 active site. 38 Telaprevir (Incivek™, Vertex Pharmaceuticals, Cambridge, MA, USA) is also a covalent peptidomimetic inhibitor of the NS3/4A enzyme, which also binds to the enzyme active site via its ketoamide anchor site. Initial drug development focused on HCV genotype 1 as this subtype is the most prevalent and less likely to respond to PEG-IFN/RBV therapy. Many agents in development are likely to be pangenotypic, however both boceprevir and telaprevir are approved for use only in genotype 1 patients. Preliminary studies show that there is efficacy with telaprevir on genotype 2 and genotype 4, but minimal effects on genotype 3. 39
The pharmacokinetic properties of telaprevir are summarized in Table 1. Relative to fasting, when telaprevir was administered with a low-fat meal (3.6 g fat) and a high-fat meal (56 g fat), the systemic exposure (AUC) to telaprevir was increased by approximately 117% and 330%, respectively. Therefore, telaprevir should always be taken with meals containing at least 20 grams of fat. Telaprevir is extensively metabolized in the liver via hydrolysis, oxidation and reduction. Multiple metabolites were detected in feces, plasma, and urine. In vitro studies indicated that CYP 3A4 was the major CYP isoform responsible for telaprevir metabolism. No inhibition by telaprevir of CYP 1A2, 2C9, 2C19, and 2D6 isozymes was observed in vitro. 37
The pharmacokinetics of boceprevir as studies in healthy subjects and the HCV population did not differ significantly between populations (Table 1). The pharmacokinetic steady state is achieved after 1 day of three times daily dosing, and while it should be administered with food, in contrast to telaprevir, a fatty meal is not required. Boceprevir is primarily metabolized via the aldo-ketoreductase pathway, and to a lesser extent undergoes oxidative metabolism by CYP 3A4/5. 38 While it is a partial substrate to hepatic CYP 3A4/5, it is considered a strong inhibitor of CYP 3A4/5 enzymes.
In addition, no dose adjustment of boceprevir or telaprevir is required in patients with renal insufficiency. No clinically significant differences in pharmacokinetic parameters were observed with varying degrees of chronic liver impairment in patients treated with boceprevir and therefore, no dosage adjustment of this drug is required in patients with cirrhosis and liver impairment. No data on patients with moderate or severe hepatic impairment (Child-Pugh grade B or C) is available for telaprevir. 37
Clinical Studies and Efficacy
Boceprevir
A large phase 2 clinical trial, HCV Serine Protease Inhibitor Therapy 1 (SPRINT-1), evaluated boceprevir in combination with RBV and PEG-IFN-2b in a genotype 1, treatment naive population. Boceprevir was tested as a 24 or 48-week therapy with concurrent PEG-IFN/RBV. Two treatment arms tested a lead-in strategy consisting of 4 weeks PEG-IFN/RBV followed by either 24 or 44 more weeks of all three drugs. The theoretical benefit to a lead-in strategy is that RBV and PEG-IFN reach steady state concentrations by week 4. Beginning a DAA at this point would limit the amount of time the DAA acts alone on the virus, thereby limiting the chance for development of resistance. The lead-in period showed that SVR could be predicted by the degree of PEG-IFN/RBV responsiveness in the initial 4 weeks. If reduction of less than 1.5 log was seen in the lead-in period, addition of boceprevir for 44 more weeks achieved better SVR rates than a 28 week course (55% vs. 28%). In addition, a lower-dose RBV regimen in combination with boceprevir and PEG-IFN-2b was inferior and associated with a high rate of viral breakthrough and a relapse rate similar to controls. 40
In the phase 3 trial, SPRINT-2 (Serine Protease Inhibitor Therapy 2), response-guided therapy was tested and all treatment arms underwent a lead-in with PEG-IFN/RBV. Those who achieved and maintained viral clearance between weeks 8 to 24 were treated for 28 weeks of total therapy, while others were treated 48 weeks total. SVR was achieved in 38% of the control group (PEG-IFN/RBV for 48 weeks), while SVR was achieved in 63% of the response guided therapy group, and 66% in the 48-week triple therapy group.
Relapse rates were 22% in the controls, and 9% in both response-guided group and full 48-week group. Importantly, while the addition of boceprevir to standard of care improved SVR rates for black participants and those with advanced fibrosis or cirrhosis, they should not be treated with response guided therapy based on the SPRINT-2 trial results. In these groups, the response was significantly better when treated for a 48-week course compared to 28 weeks. 41
RESPOND-2 (Retreatment with HCV Serine Protease Inhibitor Boceprevir and Peginteron/Rebetol) studied genotype 1 patients experiencing prior treatment failure with a similar study design as the SPRINT-2 trial, showing SVR rates of 66% in the 48 week arm, and 58% in the response-guided arm, compared to only 21% in the control group. Of note, this study was limited to patients who showed interferon responsiveness, defined as at least a 2-log decline with prior interferon therapy. This study also demonstrated that patients with poor response to the lead-in period of therapy (less than 1 log decline in HCV RNA at the end of week 4 of the lead-in period) had significantly higher resistance-associated-variants. 42
Telaprevir
Phase 1 trials established that 750 mg every 8 hours was an optimal dose, reducing HCV RNA by greater than or equal to a 4 log10 reduction, and sustaining the highest C trough at 1054 ng/ml. Despite rapid response to therapy, viral breakthrough with less sensitive viral strains confirmed telaprevir would have to be used with combination therapy. 43
The PROVE-1 study, the first phase 2 trial of telaprevir, compared triple therapy to standard PEG-IFN/RBV controls using a 12-week telaprevir treatment course, with a concurrent 12-week, 24-week, or 48-week PEG-IFN/RBV course. In the treatment groups assigned to receive less than 48 weeks of therapy, if patients did not achieve RVR or had breakthrough, they were treated for a full 48 weeks. SVR rates were 41% in the control group, and 61% and 69% in the 24-week and 48-week PEG-IFN/RBV + 12 weeks telaprevir groups, respectively.44,45
PROVE-2 was distinct from the former trial in that an RBV-free arm was tested, using telaprevir/PEG-IFN for 12 weeks. Although the RVR rate was 50% in the RBV-free group and 13% in PEG-IFN/RBV controls, SVR rates were 36% and 46%, respectively, compared to 60%-69% in the triple therapy groups. In addition, rates of relapse were more than doubled in the RBV-free group versus the PEG-IFN/RBV + telaprevir group. 46
PROVE-3 determined efficacy among patients with prior treatment-failure. Patients with well-characterized response to prior PEG-IFN/RBV therapy for at least 12 weeks were included. In addition, 13% of the patient population had cirrhosis. Key findings were that SVR rates were much higher when patients were retreated with triple therapy compared with PEG-IFN/RBV alone (52% for triple therapy group vs. 14% in the PEG-IFN/RBV group). SVR rates with 12 weeks telaprevir and 24 weeks PEG-IFN/RBV versus 24 weeks of telaprevir and 48 weeks of PEG-IFN/RBV did not differ significantly (51% vs. 53%, respectively). Also, more breakthroughs were seen in patients with genotype 1a (24%) versus 1b (11%). Interestingly, patients with cirrhosis had comparable rates of SVR compared to those with no or minimal fibrosis. Specifically, SVR rates were 53% and 45% in patients with cirrhosis versus 62% and 45% in patients with no or minimal fibrosis with 12 weeks telaprevir and 24 weeks PEG-IFN/RBV or 24 weeks of telaprevir and 48 weeks of PEG-IFN/RBV, respectively. 47
Shortening telaprevir therapy from 12 to 8 weeks decreased frequency of severe rash (>50% of body surface area), but increased rates of virologic failure (defined as meeting stopping criteria) from 5% in 12 week telaprevir to 10% in 8 week telaprevir during the PEG-IFN/RBV alone phase. Overall SVR rates were 75% in the 12-week group and 69% in the 8 week group. Rash management protocol lowered overall full treatment discontinuation rates by allowing for sequential discontinuation of telaprevir followed by RBV 7 days later, followed by PEG-IFN if necessary. 47
Phase 3 trials, ADVANCE and ILLUMINATE, confirmed the superior efficacy of telaprevir plus PEG-IFN/RBV versus PEG-IFN/RBV alone in treatment naive patients with and without cirrhosis. Patients with cirrhosis had improved SVR rates, ranging from 69%-75% in telaprevir groups compared to 44% in controls. Response-guided therapy was validated in those without cirrhosis, as patients with an extended RVR can be treated with 24 weeks of total therapy (12 weeks triple therapy followed by 12 weeks PEG-IFN/RBV) without lowering SVR rates.45,48
As a follow up to PROVE 3, the REALIZE trial was a phase 3 trial that analyzed patients with prior treatment failure with PEG-IFN/RBV. This trial also included a 4-week PEG-IFN/RBV lead-in arm. As expected, a gradient of SVR was seen in retreated patients, with relapsers (86%), partial responders (51%) and null responders (31%) showing decreasing rates of SVR. These SVR rates were significantly better that standard control. Lead-in showed no benefit in this study population, and did not appear to affect outcomes. An analysis of SVR by degree of fibrosis showed that in prior relapsers, SVR rates were equivalent across all groups. In contrast, in partial responders SVR occurred in 72% of patients with no or mild fibrosis, 56% in patients with bridging fibrosis, and 34% of those with cirrhosis. In null responders, the rates of SVR were 41%, 39%, and 14%, respectively.49,50
Concern had been expressed regarding patients who acquired resistant viral variants during treatment with protease inhibitors. The EXTEND trial followed patients from phase 2 and 3 telaprevir trials for 3 years after treatment. A reversion to non-resistant strains was seen in 89% of patients. In addition, the data showed 99% of patients had durable SVR after receiving telaprevir-based therapy. 45 While this data has not been formally published, it is likely that patients failing triple therapy would be at high risk of failure with re-treatment with similarly acting drugs due to undetectable levels of the resistant mutant in most patients.
Safety
Peginterferon and ribavirin
Side effects are seen in the vast majority of those undergoing therapy with PEG-IFN/RBV. Ability to achieve SVR depends upon compliance with medicine. Reduction or discontinuation of therapy due to side effects can significantly compromise patient outcomes. The early side effects of PEG-IFN/RBV include influenza like symptoms (fever, chills, myalgia, fatigue, and arthralgia), dermatologic problems (rash), and bone marrow suppression (neutropenia, anemia, and thrombocytopenia). The later complications include neuropsychiatric, cardiovascular, and exacerbations of autoimmune disease (Fig. 3).
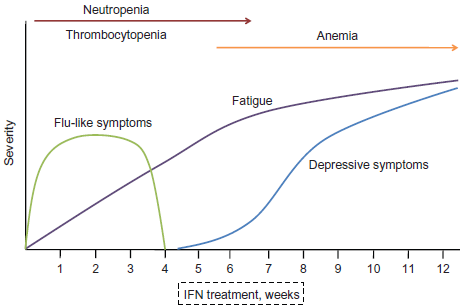
The typical onset of symptoms associated with interferon therapy begins with flu-like symptoms shortly after the first treatment, followed by progressively worsening fatigue. The onset of depressive symptoms and anxiety typically begins at 4-5 weeks of treatment and plateaus at week 12 along with the level of fatigue. Neutropenia and thrombocytopenia are seen initially, whereas low hemoglobin levels tend to persist in the latter half of the treatment period. Reproduced with permission from: Kelleher TB, Afdhal NH. Peginterferon and ribavirin for the treatment of chronic hepatitis C virus infection: Management of side effects. In: UpToDate, Basow DS (Ed), UpToDate, Waltham, MA, 2012. Copyright © 2012 UpToDate, Inc. www.uptodate.com. 54
The most common side effects are headache (54%-56%), fatigue (50%-52%), myalgias (37%-54%), and fever (22%-36%). 80 percent of patients are afflicted with at least one symptoms. Symptoms initially present within the first 48 hours of PEG-IFN administration, and only 10% of patients experience persistence of symptoms beyond the third month of treatment.31,32 Therefore patients can be reassured that symptoms improve with continued therapy and advised to take acetaminophen prior to injection of PEG-IFN (Fig. 3).
Hematologic side effects of therapy are both common and problematic. Anemia typically is seen in within the first 12 weeks of therapy, with rates of hemoglobin dropping below 10 g/dL reported between 9% and 30%.16,18,51 The etiology of the significant anemia is likely associated with the high concentrations of RBV in the erythrocyte. RBV-associated hemolysis is compounded by suppression of erythropoesis by interferon in the bone marrow. While the exact mechanism of RBV-associated hemolysis is unclear, it is associated with stabilization on dose reduction and resolution upon discontinuation. One study showed a hemoglobin drop of ≥1.5 g/dL by week 2 was an excellent early predictor for subsequent considerable hemoglobin decreases and might be used to identify candidates for early intervention against anemia in order to help maintain RBV dosing and avoid suboptimal exposure. 52 Several studies have attempted to determine if the use of erythropoietin is a reasonable strategy to limit anemia and assist in achieving SVR by limiting the need for dose reductions. Significant improvement seen in quality of life, particularly fatigue, by maintaining the hemoglobin above 12 g/dL has led many experts to adopt the use of erythropoietin as an adjunct to HCV therapy. Notably, erythropoietin has several Black Box Warnings, which include increased mortality in CKD patients, increased tumor progression and increased thromboembolic events.53,54
Neutropenia from bone marrow suppression by interferon is not uncommon. Dose reductions in PEG-IFN due to neutropenia are reported to occur in 18%-20% of patients.16,18 Severe neutropenia (ANC < 500 per uL) is reported to occur in 0.7%. 18 Despite the relatively high incidence of neutropenia, clinical important bacterial infections do not appear to occur at increased rates in those neutropenic from HCV therapy. 55 Given the lack of evidence to suggest increased risk from neutropenia, it is currently not recommended that patients receive granulocyte-colony stimulating factor (G-CSF) unless ANC drops below 500 and they are have an additional reason to be at risk of infection, ie, co-infection with HIV, cirrhosis, post-transplant immuno supression.
Severe thrombocytopenia can be seen, and usually occurs in patients with advanced cirrhosis. Platelet counts below 25,000/uL occurred at a rate of 3% in a cohort of 321 patients undergoing therapy with PEG-IFN/RBV, 56 and dose reductions were required in 3%-4% of patients secondary to thrombocytopenia.16,18 Treatment with eltrombopag, a thrombopoietin agonist, increased platelets in thrombocytopenic HCV-infected patients with advanced fibrosis and cirrhosis, enabling 95% to initiate antiviral therapy. Compared to placebo, eltrombopag allowed significantly more patients to maintain antiviral dose and achieve SVR. 57 While this is a promising agent, its use carries an increased risk of thromboembolism and the drug has a Black Box Warning for hepatotoxicity.
Depression is reported to develop in 20%-40% of patients initiating PEG-IFN/RBV, but has been reported to occur as high as 80% in one study.16,18,58 This effect has been attributed to the leading cause of decreased quality of life during treatment, which translates into dose reductions or treatment discontinuation. 58 Risk factors and baseline neuropsychiatric status are important to recognize prior to the initiation of treatment not only to assess if the patient is a candidate for therapy, but also to monitor the progress of therapy and to intervene should symptoms emerge. If patients exhibit depressive symptoms prior to treatment, initiation of an SSRI prior to PEG-IFN has proven to be beneficial. 59 Otherwise, initiation of an SSRI at the emergence of depression symptoms during treatment is considered effective at reducing symptoms and allowing patients to complete therapy. 60
Other adverse effects include a non-specific pruritic rash, which has been attributed to RBV, as discontinuation or dose reduction improves the rash. When severe, low-potency steroid cream can be applied to assist in limiting dose reduction when possible. The induction of auto-immune disease, particularly thyroid disease, has been attributed to IFN therapy. Patients may develop a variety of forms of thyroid disease including thyroid antibodies without clinical signs of thyroid disease, Hashimoto's thyroiditis, or Grave's disease. Sarcoidosis has also been reported in association with IFN in several case reports.
The safety of triple therapy
In the phase 2 study of boceprevir, SPRINT-1, the most common side effects were anemia, nausea, vomiting, and dysgeusia. Adverse events were more common in the boceprevir groups than in PEG-IFN/RBV treatment groups (11%-15% vs. 8%) and were largely due to anemia (defined as Hgb < 10 g/dL) and dysgeusia. No increase in skin or subcutaneous disorders was noted. Higher rates of anemia were noted in the boceprevir-containing regimens than in the controls. Stopping treatment for anemia was rare, however, this is likely because the use of erythropoietin was allowed in this trial. Further, the presence of anemia and erythropoietin use was associated with improved SVR in all treatment groups, 38 perhaps a reflection of increased RBV and/or boceprevir exposure. The Phase 3 trial, RESPOND-2, reported anemia (<10 g/dL) in 43 to 46% of patients exposed to boceprevir. Erythropoietin was used per protocol and less than 2% of patients discontinued therapy as a consequence. 42
Adverse events associated with telaprevir, as noted in the phase 3 trial ADVANCE, 47 were notable for an increase in rash, anemia, diarrhea, pruritis (including anal pruritis), and nausea. A severe, primarily eczematous rash (covering over 50% of body surface area) was seen in 6% of patients receiving 12 weeks of telaprevir. The rash usually resolved upon discontinuation of telaprevir, but if necessary, RBV was removed 7 days later, and subsequently PEG-IFN if there was continued progression. In total, 7%-11% of patients discontinued telaprevir due to rash and 0.5%-1.4% discontinued all study medication due to rash. Cases of Stevens-Johnson syndrome and drug rash with eosinophilia and systemic symptoms (DRESS) have also been reported. A nadir hemoglobin of less than 10 g/dL occurred in 38% of patients treated with telaprevir and 14% of controls, whereas nadir hemoglobin less than 8.5 g occurred in 9% of patients treated with telaprevir and 2% of controls. Current American Association for the Study of Liver Diseases (AASLD) guidelines recommend a decreased dose in RBV when a patient develops anemia while undergoing therapy. When telaprevir is stopped after 12 weeks, hemoglobin levels did not differ among treatment groups by 24 weeks of therapy. Erythropoietin was not used in the ADVANCE trial.
In the PROVE 2 trial, telaprevir-based groups had overall greater rates of discontinuation of therapy than the PEG-IFN/RBV-only group (21% vs. 11%). Seven percent of patients in the telaprevir group had a severe rash requiring discontinuation versus 1% of controls. Anemia was also seen at a significantly higher rate, mean 0.5-1 g/dL greater drop than controls, with return of baseline hemoglobin on drug discontinuation. Other adverse effects were pruritis, nausea and diarrhea. 46
Drug-drug interactions are an important consideration with protease inhibitors. Telaprevir and, to a lesser extent, boceprevir are substrates and strong inhibitors of CYP 3A4. Telaprevir also is an inhibitor of P-glycoprotein. Therefore, co-administration of drugs that are dependent on CYP 3A4 or P-glycoprotein for clearance and have a narrow therapeutic margin may result in toxicity. Examples include certain statins (lovastatin, simvastatin, and atorvastatin), alfuzosin, certain benzodiazepines (alprazolam, midazolam, triazolam), colchicine, eplerenone, St. John's wort, and ergot derivatives. Drugs that induce CYP 3A4 themselves may result in reduced exposure to telaprevir and boceprevir. 45 Examples include certain anti-convulsants (carbamazepime and dilantin), reverse transcriptase inhibitors (efavirenz and nevirapine), and sulfonylureas.
Viral resistance is also a reasonable concern with the addition of a DAA to PEG-IFN/RBV, as resistant mutants were observed in at least one-third of patients with poor PEG-IFN/RBV responsiveness during lead-in. Strict adherence to stopping rules, risk stratification on who to treat, and optimized dosing regimens should be used to prevent resistance patterns. Future studies are looking at combinations of complimentary drugs to improve efficacy and decrease resistance.
Patient Preference
While the benefit of achieving SVR for all patients with CHC is clear, treatment in the new era of DAAs is more intensive and complex than ever before, and the possibility of developing HCV drug resistance makes adherence a more important consideration. Furthermore, as interferon and RBV continue to be the backbone of therapy, treatment is still associated with decreased quality of life due to the occurrence of side effects in most patients. Many studies have shown that outside of clinical trials, both patient-related factors as well as treatment-related factors limit the efficacy of therapy.61–66 It is clear that patient preference is correlated with adherence to the treatment regimen which in turn greatly effects success of therapy.66–69 While the ability to achieve SVR was found to be the most important characteristic of a treatment regimen to patients, Hauber and colleagues also found that patients were willing to accept a lower probability of achieving SVR to reduce side effects of treatment. 69 Similarly, Frankel and colleagues demonstrated that the likelihood of benefit from treatment was important to their subjects. In addition, they showed that patients with more severe liver disease placed greater weight on the importance of expected benefit and less weight on the risk of side effects than patients with milder disease. 63 Other treatment-related factors such as pill burden, ease of use of injection device, and frequency of injection also improve adherence to CHC therapy.66,69,70 A study looking specifically at timing of health outcomes showed that patients with CHC have a strong preference to expedite an expected interval of poor health such as one encountered by most patients undergoing interferon-based therapy regardless of the outcome of therapy. 71 In summary, prior experience with CHC therapy illustrates that patients have strong preferences related to therapy and these preferences have a significant impact on efficacy of therapy. Consequently, their participation in treatment decisions will be key as patients and physicians weigh the risks and benefits of initiating interferon-based therapy versus deferring therapy until the approval of interferon-free regimens, especially in patients with asymptomatic and/or mild disease.
Place in Therapy
The AASLD has endorsed data-supported guidelines to assist clinicians’ approach to therapy in the face of new treatment options. Given the approval of the first protease inhibitors, there are now new standard of care recommendations for chronic HCV infection due to genotype 1. 72 The optimal treatment is now considered to be the use of boceprevir or telaprevir in combination with PEG-IFN/RBV.
For treatment-naive patients, the recommended treatment with boceprevir is a 4-week lead-in with PEG-IFN/RBV followed by 24-44 weeks of triple therapy. In those without cirrhosis, response-guided therapy should predicate treatment to 24 weeks if HCV RNA is undetectable at 8 and 24 weeks. Similarly, if telaprevir is chosen, triple therapy should commence for 12 weeks, followed by an additional 12-36 weeks of PEG-IFN/RBV, depending if viral loads are undetectable at weeks 4 and 12. Conversely, all therapy should be discontinued based on pre-defined stopping rules. For triple therapy with boceprevir, all treatment should be stopped if the HCV RNA level is > 100 IU/mL at treatment week 12 or detectable at treatment week 24. When using telaprevir in combination with PEG-IFN/RBV, all treatment should be stopped if, HCV RNA levels are >1000 IU/mL at treatment week 4 or 12 or detectable at week 24. 72
For relapsers, the same guidelines regarding triple therapy and response-guided therapy apply. However for initial partial and null responders, triple therapy with telaprevir without response-guided therapy is recommended. In addition, patients on boceprevir who continue to have HCV RNA levels > 100 IU/mL at week 12 or > 10-15 IU/mL at week 24 should discontinue all therapy given the likelihood of developing resistance. Similarly, patients on telaprevir who continue to have HCV RNA levels >1000 IU/mL at week 4 or 12 or detectable HCV RNA at week 24 should discontinue all therapy given the likelihood of developing resistance. For either DAA, should virologic breakthrough occur (> 1 log increase of HCV RNA above nadir), the protease inhibitor alone should be discontinued. 72
Regarding the IL-28B polymorphism, guidelines suggest testing should be considered when the clinician or patient desire additional indicators to aid in estimating duration of treatment or to assist in making the decision to treat at all but no specific data exist currently associating IL-28B genotype and response to triple therapy.
On the Horizon
Second generation protease inhibitors
Numerous protease inhibitors beyond telaprevir and boceprevir are in development (Table 2). Notably, TMC435 (Tibotec/Medavir/Johnson & Johnson), is a once daily dosed NS3/4A inhibitor, which has shown synergistic effects with polymerase inhibitors in vitro. 73 At week 12 EVR was recorded in 91% of those randomized to 12 weeks of TMC435 treatment, 97% in those randomized to 24 weeks of TMC435, and 69% in those randomized to the control group, respectively. Notably, viral breakthrough by week 24 occurred in 2.5% to 7.8% of the TMC435 groups, compared with 3.9% of the control group.26,74

Danoprevir (ITMN-191, RG7227) has been shown to be active against several genotypes. At week 12, 88% of those taking 300 mg danoprevir plus PEG-IFN/RBV, 89% taking 600 mg danoprevir plus PEG-IFN/RBV, and 92% taking 900 mg danoprevir plus PEG-IFN/RBV achieved undetectable viral load compared with 43% taking PEG-IFN/RBV alone. Danoprevir can be boosted with low doses of the CYP3A inhibitor ritonavir to achieve a more favorable pharmacokinetic profile, and this option is now being moved forward in ongoing studies to avoid the alanine aminotransferase increases seen in some patients treated with danoprevir. 74 Development of these newer protease inhibitors has lead to great excitement. Their ability to be combined with other DAAs allows for the potential to revolutionize treatment of CHC by eliminating PEG-IFN from the standard of care.
NS5B polymerase inhibitors
There are two new classes of drugs, the nucleoside polymerase inhibitors (NIs) and non-nucleoside polymerase inhibitors (NNIs). The NIs interact with the catalytic site for NS5B, incorporate into the elongating chain of HCV RNA, and subsequently cause chain termination (Fig. 2). NNIs bind to the NS5B protein and change the confirmation of the active site, which in turn prevents effective viral RNA synthesis.26,75
The development of the first two NIs was halted because of adverse effects. Valopicitabine (NM283, Idenix Novartis) was stopped because of adverse gastrointestinal effects and limited efficacy. R1626 (Roche) reached phase 2, but was also halted because of severe lymphopenia and visual impairment. There are currently 4 other NIs undergoing phase 2 evaluation (Table 3). Theoretically, NIs have less chance of causing resistant strains than NNIs, however at least one resistant mutation has developed for the class.26,75
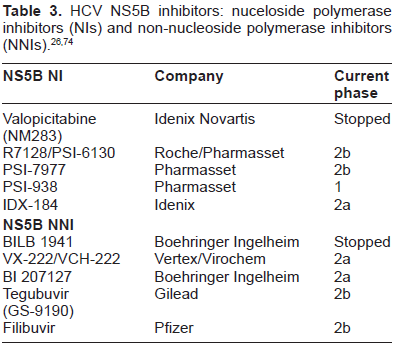
A promising NNI, filibuvir (PF-00868554, Pfizer), showed significantly reduced HCV RNA at week 4 when combined with PEG-IFN/RBV compared to PEG-IFN/RBV alone, with severity and incidence of adverse effects comparable to control groups. 76 Another NNI is entering phase 2 testing (GS-9190, Gilead), with randomization to 3 treatment arms for full 24 or 48 week treatment, depending on response to therapy.26,75 There are several other NNIs in testing (Table 3). The largest drawback for NNIs so far is the low threshold for the development of resistance. However, in combination with protease inhibitors, this is a disadvantage that may be overcome.
NS5A inhibitors
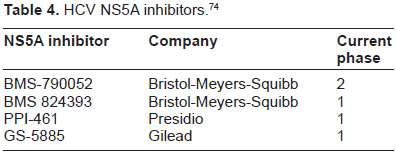
The NS5A component of the replication complex is another prime target for inhibiting HCV replication. BMS-790052 has been studied in a double blind, randomized, placebo controlled trial in combination with PEG-IFN/RBV. 75% of patients at the highest dose tested achieved an early RVR (HCV RNA < 10 IU/mL) at 4 and 12 weeks compared to 8% in the PEG-IFN/ RBV group. 58 The NS5A inhibitors have been identified as ideal candidates for combination therapy with protease inhibitors. There are 3 other NS5A inhibitors that remain in phase 1 status (Table 4).
HCV NS5A inhibitors. 74
Cyclophilin inhibitors
Cyclophilin inhibitors are a unique class of drugs with early data showing efficacy for the therapy of CHC. Cyclophilins are involved in protein folding, transport, secretion and play an important role in immune response. In vitro studies have shown that cyclophilin A and B are used by the HCV to mediate viral replication. Cyclosporine, via its inhibitory effects on cyclophilins is able to inhibit HCV replication in vitro and in vivo.26,77 While cyclosporine is not an ideal candidate for therapy of CHC due to its immunosuppressive effects, drugs that specifically inhibit cyclophilins are in development.
Alisporivir (formerly known as Debio-025) is a synthetic cyclosporine analog that inhibits the binding of the host cell protein cyclophilin A to the NS5A component of the HCV replication complex. As a result, alisporivir does not induce viral resistance and is pangenotypic in its ability to suppress HCV RNA replication. At day 29 of treatment, the PEG-IFN-2a plus alisporivir 200 mg daily arm showed a 4.8 log decline from baseline HCV RNA (51). A phase IIb trial is underway to evaluate the efficacy of this drug in combination with PEG-IFN and RBV.77,78
SCY-635 is another non-immunosuppressive analog of cyclosporine A that exhibits potent suppression of HCV RNA replication in vitro by binding to human cyclophilin A. A phase 2a clinical trial that involves SCY-635, PEG-IFN, and RBV for treatment-naive CHC genotype 1 patients is under way.26,77
Therapeutic vaccination
Therapeutic vaccination has the goal of improving viral clearance by boosting virus-specific T-cell response in the host through vaccine-mediated inoculation of specific antigens. GI-5005 is a yeast-based therapeutic vaccine containing HCV NS3 and core antigens recognized by the CD4 and CD8 T cells of persons who spontaneously clear HCV infections. GI-5005 + PEG-IFN/RBV was well tolerated and improved the SVR rate by 10% to 22% compared with controls. GI-5005 also significantly increased HCV-antigen-specific proliferation and cytotoxic cytokine secretion of peripheral blood T cells in immunized patients, indicating that it enhanced HCV-specific immunity. 26
Combination therapies
Optimal combination therapy would (1) target two or more different sites in the HCV replication cycle, (2) reduce the chance of viral resistance by achieving rapid and sustained viral undetectability, and (3) have an equal or better safety profile than boceprevir or telaprevir plus PEG-IFN/RBV. The first trial evaluating combination DAA therapy was the INFORM-1 proof-of-concept study. A total of 87 genotype 1 patients received 13 days of NS3/4A protease inhibitor danoprevir (RG7227) with RG7128 an NI versus matched placebo combination and all groups followed with PEG-IFN/RBV. Median drop in HCV viral load was 5 log for treatment naive patients, and was similar for treatment experienced patients including null responders at the end of the 2 week combination therapy. RVR rates as well as eRVR at 12 weeks were markedly increased in cohorts receiving the protease inhibitor and NI combination. No virologic resistance to either DAA was seen, and no serious adverse events were reported. 79 INFORM-SVR, 80 a phase 2b trial sponsored by Roche, is assessing the safety and efficacy of a polymerase inhibitor (RO5024048) and ritonavir-boosted a NS3 protease inhibitor (danoprevir) with and without RBV. Data is estimated to be available in August 2012.
Tegobuvir, an NNI, and GS-9256, an NS3 protease inhibitor were assessed with RBV as well as with and without PEG-IFN in genotype 1 treatment naive patients. RVR was observed in 7% (1/15) of patients receiving tegobuvir/GS-9256, 38% (5/13) receiving tegobuvir/GS-9256/RBV, and 100% (14/14) receiving tegobuvir/GS-9256/PEG-IFN/RBV. Transient elevations in serum bilirubin occurred in all treatment groups. 81
Another recent phase 2 trial investigated the antiviral effect and safety of BI 201335 (an NS3/4A protease inhibitor) and BI 207127 (an NS5B NNI) with RBV. Thirty-two treatment-naive patients with genotype 1 were randomly assigned to groups that were given 400 mg or 600 mg of the NNI 3 times daily plus 120 mg protease inhibitor once daily and 1000 to 1200 mg/day RBV for 4 weeks. In the group given BI 207127 600 mg 3 times daily, the rates of virologic response were 82%, 100%, and 100%, respectively, and did not differ among genotypes. One patient in the group given 400 mg 3 times daily had virologic breakthrough (≥1 log(10) rebound in HCV RNA) at day 22. The most frequent adverse events were mild gastrointestinal disorders, rash, and photosensitivity. 82
PSI-7977, an NS5B nucleoside polymerase inhibitor (Table 3), was administered for 12 weeks in combination with RBV without PEG-IFN elicited 100% SVR in 10 patients with genotype 2 or 3. PSI-7977 is a once daily drug whose safety and tolerability profile testing revealed no treatment discontinuation and no emergent laboratory values requiring intervention. In vitro testing has shown a similar potency profile for genotype 1 versus 2/3. Genotype 1 clinical trials with PSI-7977 plus RBV are underway and eagerly awaited.83,84
Conclusion
The recent approval of the first DAAs, boceprevir and telaprevir, is an incredible advancement in HCV therapy. Still, there are questions that remain regarding the future of HCV therapy. The most important is whether or not interferon's immune modulating effect on the host is required for a virologic cure. The evidence to support the importance of the immune effect of interferon is shown in the CC haplotype on the IL-28B gene. Yet, it may be possible that potent combinations of targeted agents will eliminate the need for immune manipulation. Both clinicians and patients eagerly await an interferon-free regimen and preliminary studies support this as an attainable goal, yet larger trials with long-term efficacy and safety remain to be completed.
HCV therapeutics is rapidly evolving. Over the next few years we anticipate the release of increasingly effective and safe agents. New treatment regimens carry the hope that reduced side effects relative to current treatments might result in higher adherence rates and better clinical effectiveness. These rapid changes make patient management a challenge. Should we continue to warehouse our patients in anticipation of better therapies or will this place them at risk for irreversible disease progression?
As with the previous standard of care of PEG-IFN and RBV, the current standard of care for genotype 1 infected patients, triple therapy combining PEG-IFN, RBV and a protease inhibitor, will leave some patients uncured. Nonetheless, we should not lose sight of the incredible improvement in HCV therapy that has just occurred. It remains that we are in an exciting and progressive time in the history of HCV therapy, and patients and physicians alike are justified in being optimistic about the near future.
Author Contributions
Wrote the first draft of the manuscript: JNG. Contributed to the writing of the manuscript: JNG, APD, NR. Agree with manuscript results and conclusions: JNG, APD, NR. Jointly developed the structure and arguments for the paper: JNG, APD, NR. Made critical revisions and approved final version: JNG, APD, NR. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.