
Abstract
Phenylketonuria (PKU) is an autosomal recessive disorder related to a deficiency in the enzyme phenylalanine hydroxylase (PAH), which converts phenylalanine to tyrosine. As a result, phenylalanine can accumulate in the bloodstream, potentially leading to severe neurologic sequelae. Traditionally, PKU management involves strict dietary phenylalanine restriction, although adherence to this diet is suboptimal, necessitating improved therapeutic options.
Sapropterin (Kuvan®) is a synthetic form of tetrahydrobioterin (BH4), a cofactor for PAH, and offers promise for patients with residual enzyme production. In four pivotal phase 2 and 3 trials, as well as several smaller trials, sapropterin has demonstrated significantly improved plasma phenylalanine concentrations in patients with BH4-responsive PKU. Furthermore, data exist to support reduced dependence on a restricted phenylalanine diet. Sapropterin has a favorable safety profile, but further studies are warranted to evaluate its long-term effects.
Sapropterin represents a significant advancement in PKU management, and its clinical role may continue to evolve as more data become available and clinicians gain experience with this novel pharmacologic agent.
Introduction
Phenylketonuria (PKU) is a hereditary metabolic condition affecting approximately 1 in 13,500 to 19,000 newborns in the US. 1 Over 500 different genetic mutations have been identified among various ethnic groups affected by PKU. 2 Overall, the incidence in the US is highest among newborns of American Indian, Alaska Native, and Northern European descent. 3 Typically, this autosomal recessive disease is related to a deficiency in phenylalanine hydroxylase (PAH), a hepatic enzyme needed to convert the essential amino acid phenylalanine to tyrosine (Fig. 1). 4 As a result of this enzyme deficiency, phenylalanine can accumulate in the bloodstream and tissues. In addition to PAH deficiency, defects in tetrahydrobiopterin (BH4), a cofactor required for PAH activity, can also contribute to elevated phenylalanine concentrations, accounting for about 2% of cases. 5 Recently, this cofactor has become an interest area for researchers, since it assists in the hydroxylation of phenylalanine to tyrosine, thereby decreasing serum phenylalanine concentrations. 6
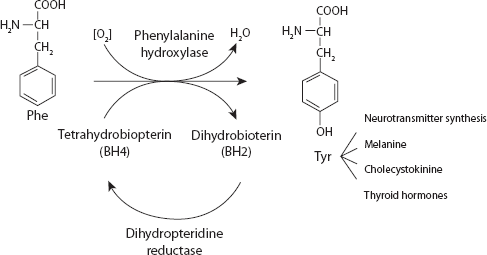
The metabolism of phenylalanine. 4 Published with permission of the editor (van Zuiden Communications B.V., the Netherlands).
Hyperphenylalaninemia (HPA), or an overabundance of blood phenylalanine, can contribute to central nervous system (CNS) abnormalities, including microcephaly, impaired brain growth, and disturbances in neurotransmitter synthesis. 5 If untreated, it can lead to irreparable neurological damage, increasing the risk for hyperactivity, gait impairments, seizures, and intellectual impairment.5,7 Individuals with elevated blood phenylalanine concentrations are also at increased risk for cognitive dysfunction, behavioral difficulties, and visual impairment, while poor metabolic control of PKU has been associated with significantly lower scores on measures of IQ, attention, and reaction time in children and adults.1,7–9
Although the exact mechanism by which HPA causes mental retardation remains unknown, the clinical implications are significant. Thus, early identification and treatment of PKU has become a focus of many clinicians. Routine screening of newborns is recommended by the US Preventative Services Task Force, with support from the American Academy of Family Physicians and the American College of Medical Genetics.10–12 HPA can be classified based on phenylalanine concentrations, with different phenotypes displaying various degrees of disease: mild HPA (150–600 μmol/L), mild PKU (600–1200 μmol/L), and classic PKU (>1200 μmol/L). 13
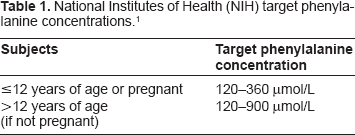
Traditionally, management of HPA has involved patients following a very restrictive diet, limiting intake of foods high in phenylalanine such as aspartame, beef, cheese, eggs, fish, pork, poultry, soy products, nuts or seeds. 14 Although these dietary measures are often the mainstay of therapy for children, a restrictive diet has also been correlated with improved cognitive function among adults.15,16 Unfortunately, compliance with this restricted diet is suboptimal; approximately 75% of patients with PKU become noncompliant. 17 Addition of amino acid supplements free from or low in phenylalanine are also important dietary measures in patients with PKU. 18 Close monitoring of phenylalanine concentrations is essential for affected individuals, and age-based target concentrations have been recommended by the National Institutes of Health (NIH) (Table 1). 1
National Institutes of Health (NIH) target phenylalanine concentrations. 1
Recently, a new pharmacologic agent has become available, providing a novel treatment option for patients with PKU. Although dietary phenylalanine restriction remains an important component of the overall management plan, many patients may benefit from BH4 supplementation. Other options, including somatic gene therapy and enzyme therapy, are currently being investigated but are not yet available in the US.
Mechanism of Action, Metabolism, and Pharmacokinetic Profile
Mechanism of action
Sapropterin (Kuvan®), 6R-tetrahydro-L-biopterin or 6R-BH4, is a synthetic form of BH4 and indicated for the treatment of HPA in patients with BH4-responsive PKU in conjunction with a phenylalanine-restricted diet. 19 BH4 is a cofactor for the enzyme PAH and enhances the hydroxylation of phenylalanine to tyrosine through an oxidative reaction. 17 Since levels of PAH are absent or deficient in patients with PKU, treatment with BH4 can promote the metabolism of phenylalanine and potentially lower phenylalanine concentrations. Sapropterin is the first pharmacological chaperone drug to be approved. Pharmacological chaperones are small-molecule chemical compounds that stabilize the target protein conformation or restore the function of affected biological pathways. BH4 corrects protein misfolding by decelerating protein aggregation, reducing hydrophobicity and slowing degradation. 20 The PAH enzyme pathway has been studied as a potential target for therapy, and an important advance in the management of PKU has emerged.21,22 Although initial versions of exogenous BH4 included a mixture of 6R-BH4 and 6S-BH4, sapropterin includes only the biologically active component, 6R-BH4.6,23,24 This agent has been shown to decrease serum phenylalanine concentrations in patients with mild HPA or PKU who maintain some residual PAH function.
Metabolism
Patients with hepatic impairment should be monitored when receiving sapropterin. Hepatic damage has been associated with impaired phenylalanine metabolism.19,25 However, the effects of hepatic or renal impairment on pharmacokinetics of sapropterin have not been studied.
Pharmacokinetic profile
The pharmacokinetic profile of 6R-BH4 has been described in several studies, with results summarized in Table 2. Overall, this agent has exhibited a rapid absorption and distribution phase, as well as a long elimination phase. In one study evaluating four healthy adults receiving a single oral dose of dissolved 6R-BH4 (10–20 mg/kg), elimination kinetics were slightly faster at higher plasma concentration. 26 One subject received both a 10 mg/kg and 20 mg/kg dosage, demonstrating a longer elimination half-life with the higher dose (T = 5.1 versus 4.2 hours, respectively). The area under the curve (AUC0–10 h) was 1.6 times higher in the 20 mg/kg dose as compared to the 10 mg/kg dose (3046 versus 1958 nmol*h/L). Although sublingual formulations of sapropterin are not commercially available, this study did compare subjects receiving oral and sublingual administration of the same dose of BH4, and plasma concentrations were 58%–76% higher with sublingual administration, although an explanation for this difference is not clear.
Summary of trials evaluating pharmacokinetics of sapropterin.
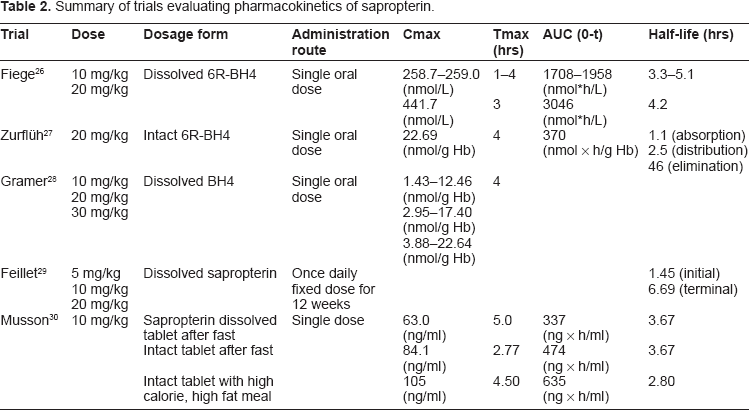
Another study by Zurflüh and colleagues examined 71 subjects with mild HPA (n = 35), mild PKU (n = 19), and classic PKU (n = 17). 27 Consistent with pharmacokinetic data in healthy adults, this study demonstrated a rapid absorption and distribution phase, followed by a long elimination phase. Although the maximum concentration was achieved 4 hours after BH4 administration, a delayed pharmacological response was observed, as shown by a reduction in phenylalanine concentrations 8 to 24 hours after each dose.
More recently, a study by Gramer and colleagues examined the drug concentrations among 17 subjects with mild, moderate, and classical PKU who received BH4 loading doses of 10 mg/kg, 20 mg/kg, and 30 mg/kg. 28 The levels of biopterin and pterin (B + P), two specific indicators of drug concentration, increased significantly with higher doses of BH4, although no relationship was observed between the B + P level and the decrease in phenylalanine concentration. Loading doses of 10 mg/kg resulted in substantially lower B + P levels in men as compared to women, although gender did not have a significant influence on the B + P levels in the 20 mg/kg or 30 mg/kg doses. Other pharmacokinetic data were consistent with findings in previous studies and did not appear to be affected by gender, phenotype, or genotype.
Once daily administration of sapropterin is supported by a study evaluating subjects receiving sapropterin at a dose of 5 mg/kg/day, 10 mg/kg/day, or 20 mg/kg/day. 29 In addition, one study assessed the bioavailability of sapropterin administered as an intact or dissolved tablet, as well as the effect of food on bioavailability. 30 Participants received a dose of 10 mg/kg as a dissolved tablet after fasting, intact tablet after fasting, or an intact tablet with a high-calorie and high-fat meal. The calculated plasma BH4 concentrations were higher with intact tablets versus dissolved tablets and were also higher during fed conditions compared to fasting conditions. Therefore, it is appropriate to administer sapropterin as a dissolved tablet with food at the same time each day. 19
Efficacy
Data are available from four pivotal phase 2 and 3 clinical trials evaluating sapropterin therapy, and results of these trials are summarized in Table 3. In addition, post-marketing studies have further examined its role in clinical practice, including its effects on dietary phenylalanine restriction.
Summary of clinical trials evaluating sapropterin therapy.
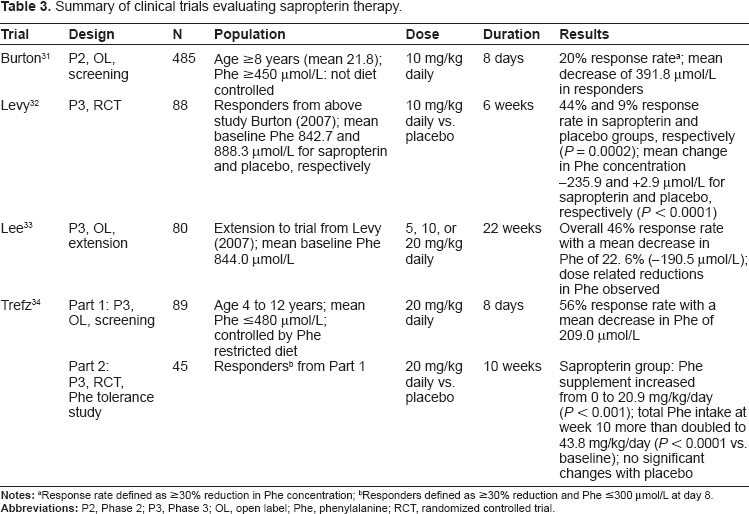
Responders defined as ≥30% reduction and Phe ≤300 μmol/L at day 8.
Pivotal clinical trials
In a phase 2, multi-center screening study, Burton and colleagues evaluated a short course of sapropterin in 485 individuals with PKU. 31 Participants designated as responders to therapy, as defined by a ≥30% reduction in phenylalanine concentrations, were eligible to participate in a subsequent phase 3 trial. Eligible subjects were ≥8 years of age and were previously nonadherent to a restricted phenylalanine diet. In addition, a previously documented phenylalanine concentration of >360 μmol/L was required, as well as a phenylalanine concentration of ≥450 μmol/L at the screening visit. Participants were excluded if they had primary BH4 deficiency, a seizure disorder, insulin-dependent diabetes mellitus, serious neuropsychiatric illness, or elevated alanine aminotransferase. Participants taking concomitant medications including corticosteroids, methotrexate, levodopa, vaccines, or any investigational agent used within 30 days of screening were excluded, along with women who were pregnant, breast feeding, or trying to become pregnant.
Study participants received sapropterin dihydrochloride 10 mg/kg by mouth once daily for 8 days and were instructed to maintain their usual dietary intake throughout the study. Upon completion of the study, twenty percent (96/485; 95% CI 16%–23%) of subjects were classified as responders to sapropterin, with a mean decrease in phenylalanine concentration of 391.8 (SD ± 185.3) μmol/L compared to baseline. Of note, significant variability existed in individual response based on baseline phenylalanine concentration, although the highest response rate was generally observed among those with lower baseline phenylalanine concentrations (response demonstrated in 54% of subjects with phenylalanine concentration <600 μmol/L). Despite an overall response rate which was lower than anticipated, a sufficient number of subjects responded to sapropterin and were candidates for a follow-up phase 3 study to evaluate the safety and efficacy of sapropterin in individuals with a previous response to sapropterin.
In this subsequent randomized, placebo-controlled study, Levy and colleagues compared 6 weeks of sapropterin 10 mg/kg orally once daily to placebo in subjects previously considered responders to sapropterin. 32 Inclusion criteria included a blood phenylalanine concentration of ≥600 μmol/L (later amended to ≥450 μmol/L) at the screening visit, age ≥8 years, and willingness and ability to comply with study procedures and to adhere to current diet. At baseline, subjects in the sapropterin and control groups had a mean phenylalanine concentration of 842.7 μmol/L and 888.3 μmol/L, respectively. Overall baseline characteristics were similar between groups, except for a higher proportion of males in the sapropterin group (66% males versus 51% males in placebo group). Baseline phenylalanine concentrations were measured 1 and 2 weeks prior to randomization, and follow-up measurements were performed at weeks 0, 1, 2, 4, and 6 of the study. Medication adherence was similar between groups, with 82% of subjects taking all doses correctly throughout the 6 week study period.
Among the 88 subjects to complete the study, individuals receiving sapropterin demonstrated a greater reduction in phenylalanine concentrations from baseline as compared to placebo (-235.9 μmol/L versus +2.9 μmol/L, P < 0.0001). Forty-four percent of individuals allocated to receive sapropterin demonstrated a ≥30% reduction in blood phenylalanine from baseline, compared to 9% of those receiving placebo (P = 0.0002). Although study participants had previously demonstrated a response to sapropterin in a phase 2 study, only 44% of subjects receiving sapropterin demonstrated a similar response in this follow-up trial, suggesting a potential for lessened response over time. Furthermore, dietary factors may have contributed to the altered response, and the relatively small sample size and unpredictable response among participants make interpretation of these results challenging.
To further evaluate sapropterin's efficacy and long-term safety, an open-label, 22-week extension study was conducted to evaluate individuals with PKU who were enrolled in the previous two studies. 33 In addition to inclusion criteria in the phase 2 and phase 3 trials described above, participation was limited to subjects who received at least 80% of the scheduled doses in the phase 3 study by Levy and colleagues. Subjects were excluded if they had discontinued the previous study for any reason other than high plasma phenylalanine concentrations or if they were expected to receive vaccinations or experimental medications during the evaluation period. Additional exclusion criteria included pregnancy, lactation, concurrent medical conditions or diseases, and/or the use of dihydrofolatereductase inhibitors, levodopa, or other medications that could potentially interfere with the study. As with previous trials, all study participants were instructed to continue their existing diet regimens during the study period.
This study was divided into three phases, beginning with a 6-week forced dose titration phase, during which participants were given sapropterin in two-week sequential courses of 5, 20 and 10 mg/kg/day. Next, a 4-week dose-analysis phase evaluated sapropterin 10 mg/kg/day among all participants. During the final 12-week fixed dose phase, subjects received doses of 5, 10, or 20 mg/kg/day based on phenylalanine concentrations measured at weeks 2 and 6 during the initial forced-dose titration phase. Participants with higher baseline phenylalanine concentrations received the higher doses of sapropterin.
In the initial forced dose-titration phase, a reduction in baseline plasma phenylalanine concentrations was observed with all three sapropterin dosages; higher doses were correlated with a greater reduction in phenylalanine concentrations. Among the 80 participants enrolled in the 22 week study, 46% achieved a ≥30% reduction in phenylalanine concentration compared to baseline, with a mean reduction of 22.6%. Although this open-label study included only known responders to BH4, the response rate is consistent with results from previous trials. Overall, sapropterin doses of 5–20 mg/kg/day demonstrated a sustained decrease in plasma phenylalanine concentration over 22 weeks in BH4-responsive individuals with PKU.
Based on data from these phase 2 and 3 trials, sapropterin was approved for the treatment of BH4-responsive PKU in 2007. Subsequently, a phase 3 randomized, double-blind, placebo-controlled, multi-center trial evaluated the efficacy and safety of sapropterin 20 mg/kg/day in increasing phenylalanine tolerance while maintaining blood phenylalanine concentration. 34 Eligible participants included children age 4 to 12 years with a diagnosis of PKU with PAH deficiency and an estimated phenylalanine tolerance of ≤1000 mg/day. A phenylalanine restricted diet was also required, with mean blood phenylalanine concentrations ≤480 μmol/L over the 6-month period leading up to study enrollment. Exclusion criteria included history of organ transplant, use of investigational agent within 30 days prior to screening, elevated serum alanine aminotransferase levels, concurrent conditions which could interfere with participation, concomitant treatment with levodopa or any medication inhibiting folate synthesis, and a history of primary BH4 deficiency.
The study was conducted in two components, with Part 1 screening for sapropterin responsiveness and Part 2 examining the safety of sapropterin and its ability to increase phenylalanine tolerance and reduce blood phenylalanine concentration. During Part 1, subjects with PKU and restricted dietary phenylalanine intake were given open-label sapropterin 20 mg/kg/day once daily for 8 days. Those who exhibited a ≥30% reduction in phenylalanine concentrations between day 1 and day 8, along with a blood phenylalanine concentration of ≤300 μmol/L on day 8, were eligible to proceed to Part 2. Following a washout period of ≥1 week, these subjects were randomized to receive a 10-week course of sapropterin 20 mg/kg/day or placebo once daily. Participants were instructed to maintain a controlled, phenylalanine-restricted diet throughout the study and a detailed food diary for 3 days of each week. Using a defined algorithm, phenylalanine supplements were added or removed every 2 weeks, beginning at week 3, based on phenylalanine concentrations from the previous week. Nutritionists and other study personnel reviewing diet diaries and adjusting phenylalanine supplements were blinded to the treatment group. Any participant with a phenylalanine concentration ≥1200 μmol/L on two consecutive weekly blood samples was instructed to stop the study drug and receive dietary counseling. Sapropterin and phenylalanine supplements were discontinued at week 10, and a final follow-up safety visit was performed at week 14.
Of the 89 subjects who completed Part 1 of the study, 50 (56%) were classified as responders, with a mean decrease in phenylalanine concentration of 64%; 45 of these responders progressed to Part 2, with 33 subjects receiving sapropterin and 12 receiving placebo. Subjects receiving sapropterin tolerated a mean phenylalanine supplement increase of 20.9 mg/kg/day (95% CI 15.4 to 26.4, P < 0.001 versus baseline), while total phenylalanine intake at week 10 reached 43.8 mg/kg/day (versus 16.8 mg/kg/day at baseline, P < 0.0001). In contrast, the placebo group tolerated a mean phenylalanine supplement increase of only 2.9 mg/kg/day, and total phenylalanine intake reached 23.5 mg/kg/day (vs. 16.3 mg/kg/day at baseline), with neither outcome achieving statistical significance. At week 10, the mean phenylalanine concentration in the sapropterin group was 340 (SD ± 235) μmol/L, compared to 461 (SD ± 235) μmol/L in the placebo group. After adjusting for potential confounders, the phenylalanine intake tolerated by the sapropterin group was significantly higher than in the placebo group, with a treatment difference of 17.7 mg/kg/day (95% CI 9 to 27, P < 0.001).
Although this study showed a better initial response rate compared to previous trials, this could be related to more selective inclusion criteria and a higher sapropterin dose of 20 mg/kg/day. Overall, this trial demonstrated that daily dosing of sapropterin 20 mg/kg/day can significantly improve dietary phenylalanine tolerance, more than doubling the mean tolerated phenylalanine intake while maintaining blood phenylalanine concentrations <360 μmol/L in subjects who were known responders.
Additional studies
Since FDA approval of sapropterin, several smaller studies have recently examined the role of sapropterin in clinical practice, specifically responder rates, effect of genotype and baseline phenylalanine concentration, and the potential for diet liberalization.
A 62% response rate was observed among individuals with classical or variant PKU (phenylalanine 401–1199 μmol/L) who were taking sapropterin 20 mg/kg/day. 35 Response rates for those with classical and variant PKU were 26.7% and 100%, respectively. Furthermore, responders were able to significantly liberalize their diet, with two subjects transitioning to an unrestricted protein diet.
Additional long-term data was provided by a small study which evaluated six children (age 5 to 12 years) who were known responders to sapropterin and had previously well-controlled blood phenylalanine concentrations. 36 Subjects demonstrated improved phenylalanine tolerance while maintaining mean plasma phenylalanine concentrations at <360 μmol/L throughout the 2-year study period.
In another small long-term study, 14 of 16 responders to sapropterin were able to achieve long-term blood phenylalanine control, with a mean treatment duration of 56 months (range: 24 to 110 months). 37 Of these responders, seven successfully eliminated dietary phenylalanine restriction, while the remaining seven were able to increase phenylalanine intake from 200–300 mg/day to 800–1000 mg/day.
Due to the variations in response to sapropterin, associations between genotype and response are being investigated. One study by Elsas and colleagues examined the response to sapropterin in patients with known classic PKU on a restricted diet. 38 The frequency of severe mutant PAH alleles was higher in the non-responder group (67%) compared to the responder group (40%). However, response could not be consistently predicted based on genotype.
Another study by Trefz and colleagues reviewed 250 PKU cases and the correlation between genotype and BH4 responsiveness, revealing an association between genotype and non-responsiveness. 39 However, many inconsistencies existed, including variable responses among patients with the same genotype. The authors concluded that these inconsistencies could be in part due to external factors such as intestinal absorption of BH4, catabolic conditions, or other genetic factors. Further studies are needed to determine genotype-phenotype correlation to BH4 responsiveness.
Overall, these small studies provide additional support for sapropterin in clinical practice and offer promise to clinicians managing PKU. Based on available data, it appears that select individuals may exhibit sustained response to sapropterin and may be able to consider a liberalized diet when taking this agent. Although response is not yet predictable, a trend may exist between response and individual genotype.38,39 However, other factors are likely relevant and should be considered when making treatment decisions.
Safety
Published clinical trials evaluating sapropterin suggest a favorable safety profile for this agent. The most commonly reported adverse events include headache (10%–20%), nasopharyngitis (15%), pharyngo-laryngeal pain (2–14%), upper respiratory tract infection (3%–28%), vomiting (2%–13%), diarrhea (5%–10%), and abdominal pain (5%–9%).31–34 Post-marketing studies have also demonstrated good tolerability of sapropterin in clinical practice.35–38 However, long-term safety data are not yet available, and further studies are needed to evaluate its effects in young children, pregnancy, and renal or hepatic impairment. Appropriate monitoring is warranted in these special patient populations.
No human studies have evaluated potential drug interactions with sapropterin, although theoretical interactions do exist.19,25 Medications which affect folate metabolism, such as methotrexate, may inhibit the enzyme dihydropteridinereductase (DHPR), thus potentially decreasing BH4 concentrations. These medications should be used cautiously in patients taking sapropterin. BH4 may induce nitric oxide-medicated vasorelaxation, so concomitant use of medications such as phosphodiesterate-5 inhibitors (including sildenafil, vardenafil, or tadalafil) should be avoided due to the additive vasorelaxation potential. Post-marketing surveillance in patients without PKU taking sapropterin and levodopa concomitantly has indicated a potential increase in neurological disorders, such as convulsion, over-stimulation, and irritability. Therefore, this combination of medications should be used cautiously.19,25
Patient Preference
Patients affected by PKU encounter numerous challenges, including the high cost of treatment and the need for a strict ‘diet for life’. Several factors challenge compliance with this diet, including time constraints, social pressure, and dissatisfaction with restrictions. 40 Thus, the need for an alternative approach to management of PKU has become apparent. As the first oral pharmacologic agent available for PKU, sapropterin offers promise to many individuals facing these challenges.
One important consideration for many individuals affected by PKU is the high cost of treatment. On average, the annual cost of dietary supplementation and monitoring can approach $57,000. 41 Since insurance companies do not always cover the cost of sapropterin, expenses for individual patients can be significant. In order to improve affordability of this product, BioMarin has created the BioMarin Patient and Physician Support (BPPS) Program to assist patients in obtaining the drug from a specialty pharmacy, regardless of insurance or income status. 42 Since dietary supplementation and phenylalanine monitoring are still required for most patients taking sapropterin, the overall cost of management can be burdensome despite the availability of this support program.
Potential benefits have been reported among patients attempting to ease dependence on a reduced phenylalanine diet.43,44 In several studies, participants taking sapropterin were able to gradually add phenylalanine-containing products to their diet.34–37,45 Tolerance to dietary adjustment may be assessed by gradually adding milk and/or egg powder to the diet, although close monitoring of blood phenylalanine concentrations is warranted during this time period. Further studies are needed to examine the potential role of sapropterin in easing dietary restriction, but this agent offers promise to patients who struggle with dietary adherence.
Place in Therapy
Currently, sapropterin is indicated for the treatment of BH4-responsive PKU as an adjunct to a reduced phenylalanine diet. 19 However, despite its availability in the US since 2007, limited data are available evaluating the long-term safety and efficacy of sapropterin. Furthermore, although this agent may offer promise for select individuals who respond to therapy, identification of appropriate candidates for sapropterin may be challenging for clinicians. In clinical studies, response to sapropterin is neither consistent nor predictable, and some subjects in phase 2 and 3 trials have demonstrated that the effect of sapropterin may not be sustained. The variability in response may make use of this agent less appealing for some clinicians.
The manufacturer's recommended starting dose for sapropterin is 10 mg/kg once daily, which may be adjusted monthly based on phenylalanine concentrations. 19 Additionally, specific approaches to determine responsiveness to sapropterin have been proposed. 45 Dietary consistency is recommended when initiating therapy, since compliance with diet may influence response. Patient genotype may help predict response to BH4 therapy, but many inconsistencies exist. Future studies are needed to evaluate the phenotype-genotype correlation in patients with PKU. 39 Phenylalanine blood concentrations should be monitored to assess response to drug therapy. Patients should be initiated on 10 mg/kg/day dosing and phenylalanine concentrations checked one week after initiating therapy and then periodically for one month. If phenylalanine concentrations have not decreased from baseline after one month, the dose may be increased to 20 mg/kg/day. Patients whose phenylalanine concentration does not decrease after one month are non-responders and should discontinue treatment. 19 Other sapropterin challenge protocols have been evaluated, including a 24- or 48-hour challenge with sapropterin 20 mg/kg initiated on day 1, followed by phenylalanine concentration measurements 8, 16, and 24 hours after the dose. If a <30% reduction in phenylalanine is observed, a second day of sapropterin 20 mg/kg is administered, followed by the same measurements performed on day 1. If a <30% reduction in phenylalanine concentration is observed on day 2, the patient is considered a non-responder. If a ≥30% reduction in phenylalanine concentration is observed on either day, the dose may be adjusted between 5–20 mg/kg/day to maintain the phenylalanine concentration in the desired therapeutic range. 46 In addition to this proposed approach, various other methods have been utilized to determine responsiveness. Primarily, the differences among these approaches are related to initial dosing and the duration of sapropterin challenge.
Sapropterin is available as a 100 mg tablet and should be dissolved in 4 to 8 ounces of water or apple juice. 19 Since the typical maintenance dose is 5–20 mg/kg once daily, pill burden may be an issue for some patients.
Most published studies have assessed surrogate outcome measures such as serum phenylalanine concentrations, demonstrating improved control of PKU. Unfortunately, no results are available to demonstrate a clear benefit in neurological development or other clinical sequelae. Specifically, the impact on neurological outcomes or quality of life remains unknown. Typically, a response to treatment has been defined as a reduction in blood phenylalanine of at least 30%, although this is not necessarily correlated to target concentrations recommended by the NIH.
Studies are needed to further evaluate the role of sapropterin in maternal PKU. Poor control of blood phenylalanine concentrations in pregnancy can result in mental retardation, facial dysmorphism, microcephaly, intrauterine growth retardation, developmental delay, and congenital heart disease.47,48 Restricted protein intake can also interfere with fetal growth. 49 Although the use of sapropterin in maternal PKU has been reported, no controlled trials have evaluated the use of sapropterin in pregnancy, and the agent is classified as pregnancy category C.19,50 One study examining the outcomes of maternal PKU demonstrated similar outcomes among non-PKU pregnancies and individuals with PKU who maintained phenylalanine concentrations between 120 and 360 μmol/L during pregnancy. 48 It is recommended that women receive preconception counseling and achieve goal phenylalanine concentrations before pregnancy. 51 Furthermore, a pregnancy registry has been developed to monitor pregnant women with PKU and their offspring. 19 Due to the need for an agent which can improve phenylalanine control and/or ease dietary restriction in pregnant patients with PKU, further studies are needed to clarify the risks and benefits of sapropterin in maternal PKU.
Other treatment modalities are currently being studied, offering promise to patients with PKU. Oral large neutral amino acids (LNAA) may have a role in competitively inhibiting phenylalanine, with one double-blind, randomized, placebo-controlled trial demonstrating a significant decrease in blood phenylalanine concentrations by 39% from baseline. 52 However, LNAA therapy is not currently available in the US, and its role in PKU remains unclear, particularly with regard to concomitant use of dietary restriction or sapropterin therapy. Somatic gene therapy and enzyme therapy are also being studied but have not yet emerged as effective treatment options for PKU.53–56 PEGylated recombinant phenylalanine ammonia lyase (PEG-PAL) decreases phenylalanine levels by converting phenylalanine to insignificant levels of ammonia. Based on pre-clinical data, a Phase 1 clinical trial has been initiated with PEG-PAL for the treatment of PKU. 57 Although a liver transplantation represents the most definitive therapy for PKU, the potential risk does not justify it as an appropriate substitute for dietary restriction. 58
Other considerations, such as vitamin deficiencies or osteoporosis, must also be made when managing patients with PKU, regardless of whether a patient is taking sapropterin. Restriction of dietary protein may necessitate supplementation of vitamin B12, calcium, vitamin D, amino acids, or other micronutrients as appropriate.
Conclusions
Sapropterin represents the first FDA-approved medication for PKU, offering promise for many patients struggling with a strict lifelong diet. Although dietary phenylalanine restriction and amino acid supplementation remains a standard approach when managing this condition, a trial of sapropterin is appropriate in patients with PKU in order to assess response to therapy. It is not possible to predict response among patients, but clinical trials have demonstrated that the role is best defined in those with baseline phenylalanine concentrations >450 μmol/L. Phase 2, phase 3, and post-marketing studies have demonstrated a benefit in reducing blood phenylalanine concentrations among patients who respond to sapropterin therapy, as well as improving dietary tolerance of phenylalanine. However, the role of this agent may be limited due to its high cost and the lack of long-term safety and efficacy data. Further studies are needed to elucidate its role, particularly the ability to ease dietary restriction, to treat maternal PKU, and to be used in combination with LNAA therapy or other future treatment modalities.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.