
Abstract
Over a decade has passed since several groups identified the chemokine receptors CXCR4 and CCR5 as key co-receptors for HIV entry. CCR5 is more important in HIV transmission and during the early course of HIV infection. It is also apparent that protection from HIV infection is afforded to those lacking CCR5–-the so called delta-32 homozygotes; in those heterozygous for this mutation, an attenuated course of HIV-infection is observed. Provocatively, those with modified expression of CCR5 are physiologically normal with the exception of poorer outcomes with some of the viral encephalitides specifically West Nile virus and Tick Borne encephalitis. The small molecule, orally-bioavailable CCR5 receptor antagonists, including, maraviroc (MVC), are allosteric inhibitors that lock the CCR5 receptor into a conformation such that the receptor is not able to bind HIV envelope protein; the molecules also variably block intracellular signalling induced by different receptor-binding chemokines.
The aims of this review on the CCR5 receptor inhibitors are to summarise information relevant to treatment in individuals with HIV-1 infection. Data from the licensing studies, the side-effect profile and putative long-term risks of CCR5 receptor inhibitor exposure, tropism testing and mechanisms of resistance will be reviewed. The potential for using this class of agent as an immunomodulating agent will be detailed. Given that MVC is the only licensed drug in this class at present and reflecting the greater body of work describing this agent, the majority of information in this review relates to MVC. Last, the authors propose the place of MVC in the hierarchy of HIV therapy and future opportunities for research.
Introduction
Control of viral replication to below the level of quantification using life-long continuous 1 combination antiretroviral therapy (cART) has radically changed HIV infection and its sequelae from an invariably fatal to a chronic illness.2–4 Some of the success of cART has been the development of new classes and new drugs within old classes of antiretroviral therapy. These new drugs are potent and have favourable side-effect profiles 5 although long-term exposure to some agents appears associated with toxicity costs.6–8
However, while morbidity and mortality has been reduced, life expectancy is still not normalized compared to an aged-matched population. 9 Increasingly, a number of conditions additional to the traditional AIDS-defining conditions 10 that impact on morbidity and mortality–-collectively referred to as serious non-AIDS (SNA)11–13–-are recognised; examples include cardiovascular disease and non-AIDS cancers. Cohort data have demonstrated that there is a continuum of risk for AIDS and SNA even across CD4+ T-cell count strata; risk being lowest in those with the highest CD4+ T-cell count. 14 Cohort data also suggest that normalisation of the CD4+ T-cell to above 500 cells/μL and starting cART at higher CD4+5,14–19 improve long-term survival. However, there are unmeasured confounders which may influence these cohort data; randomised data to more precisely define the risk benefit of starting cART at CD4+ >500 cells/uL vs. deferral to 350 are being gathered. 20
The pathogenesis of HIV and some of its sequelae including SNA is incompletely understood but chronic inflammation is thought to play a central role. Data from the SMART study showed, that higher levels of inflammatory biomarkers (hsCRP, interleukin-6 and the D-Dimer) 21 were an independent risk factor for all-cause mortality and rebounds in these biomarkers was significantly greater in those ceasing antiretrovirals as part of the randomised strategy. Moreover, while inflammatory biomarkers are lower in people on cART or among the so called elite controllers, they are not normalized.22,23 Whether inflammatory biomarkers are causal or surrogates for a dysregulated immune response is uncertain. There are many potential drivers of inflammation in the setting of HIV, include bacterial translocation across a damaged gut,24,25 low level HIV replication in compartments which are difficult for many antiretroviral agents to penetrate 26 and “latent” infections such as those of the Herpes group viruses (reviewed in 27 ). As a consequence there is interest and research exploring strategies to reduce immune activation including intensification with antiretroviral agents such a raltegravir28,29 and maraviroc.30,31
Despite the positive impact of cART on HIV-related outcome, there are still some caveats to the long-term use of cART. To date, no combination of antiretroviral agents has been able to eradicate HIV infection32–35 necessitating, for the most part, life-long therapy. In addition, immune dysregulation and activation are reduced but not normalized by cART24,25,27,36 and immune restoration in some patients with adequate control of HIV replication remains suboptimal.13,14,37–39 Moreover, the duration of successful cART may be limited by adherence fatigue, resistance and/or cumulative antiretroviral-related toxicities resulting in increasing morbidity and mortality from ischaemic heart disease and stroke.6–8
Maraviroc, the first-in-class licensed CCR5-receptor blocker, 40 targets a host receptor critical for HIV virus entry into CD4+ T-cell and monocyte/macrophages and hence has a very different mechanism of antiviral activity from all other antiretroviral agents. This agent has had, until recently, a dual pathway of development, on the one hand as therapeutic agent for the treatment of HIV-infection and on the other hand, as an anti-inflammatory agent in the setting of rheumatoid arthritis (RA). There is considerable interest in understanding better the potential of maraviroc not only an antiviral agent but as an immunomodulating/inflammatory agent in the setting of HIV-infection.
Chemokine-Receptor-5 (CCR5) Receptor Biology
Over the past two years, our understanding of chemokine-receptor biology has advanced substantially. Protective and pathological immune responses are dependent on a coordinated immune response. One of the mechanisms involved in leucocyte trafficking is mediated via chemokine receptors. These are host cellular receptors, whose ligands are chemokines, small low molecular weight cytokines, which, as indicated by their name, promote movement of cells under a chemotactic gradient. 41 Both CCR5 and CXCR4 are structurally related and are members of the seven-transmembrane G-protein coupled receptors (GPCRs) superfamily.42,43 There is a common 7-transmembrane structure, with extra and intracellular loops; some of the difficulties in defining their crystal structure has been due to the hydrophobic nature of the proteins, 44 particularly in the regions that span the membrane. Ligand bound chemokine receptors undergo rapid phosphorylation (there are 7 phosphorylation sites in CCR5) 45 and following this, a conformational change ensues, such that the cytoplasmic domains are able to couple with intracellular heterotrimeric G protein (reviewed in 46 ). The disassociation of the receptor bound G-protein allows secondary and tertiary intracellular signals initially through phospholipase C and subsequently protein kinase C.47,48 B-arrestins play an important role in the attenuation of signaling, first by limiting further G protein coupling and second, by facilitating the binding of the receptor to clarthrin allowing receptor internalization. 49 The receptors are recycled to the cell surface or enter a degradative pathway which is ubiquitin controlled. 50
Many chemokine receptors including CCR5 are promiscuous, binding more than one chemokine, with some chemokines acting as agonists and others binding but not triggering receptor signalling effectively (e.g. CCL7) thus acting as antagonists. 51 In contrast, there is only a single defined agonistic ligand of CXCR4, CXCL12 (previously called stromal-derived factor 1). CXCL12 is highly conserved between species, with 6 isoforms identified. 52 Absence of the CXCR4 receptor or CXCL12 are lethal in utero in mice, with defective vascular, cardiac and haematopoietic development associated with the former and deficient B-lymphopoiesis and myelopoiesis and abnormal neuronal and cardiovascular development in the latter.53,54
The CCR5 receptor mediates chemotaxis by the CC-chemokines CCL5 (previously called RANTES), CCL3 (previously called MIP-1α) and CCL4 (previously called MIP-1β); other chemokines that can bind CCR5 include CCL7, CCL8, and CCL13; CCL13 is thought to function as an antagonist as described above. The chemokine receptor, CCR5, is upregulated during inflammatory conditions and is thought to play an important role in recruitment of Th1 cells to the site of inflammation and/or infection. The CCR5 receptor is expressed by antigen-presenting cells (monocytes, macrophages, immature dendritic cells, Langerhans cells), immune effector cells,55,56 human TReg- (reviewed in 57 ) gastric associated lymphoid tissue (GALT), plasmacytoid dendritic cells (reviewed in 58 ) and natural killer cells.
Recognition of the Critical Importance of CCR5 as a Co-receptor for HIV Entry into the CD4+ T-cell
In 1995 Cocchi et al 59 reported that MIP-1alpha (CCL3), MIP-1beta (CCL4), and RANTES (CCL5) when applied together could prevent HIV entry into host cells. In 1996 several groups identified the chemokine receptors CXCR4 and CCR5 as key co-receptors for HIV entry; 60 other chemokine receptors have also been identified that can facilitate HIV infection in vitro 61 but CXCR4 and CCR5 appear to be the critical co-receptors for HIV infection in vivo. CCR5 using viruses (R5 viruses) overwhelmingly dominate at transmission and are the major strain of virus found early in the course of HIV infection with an estimated 80% of untreated patients having R5 strains; 62 moreover there appears to be a selection pressure on the virus to remain R5-tropic following seroconversion that is maintained for several years, the underlying mechanism of which is unclear. However, one possibility is that the production of CXCL12 by dendritic cells (DC) resulting in down-regulation of CXCR4 may delay the switch to X4 virus.63,64 Another hypothesis regarding the source of this selection pressure includes competing pressures between co-receptor availability, the action of neutralising antibodies, and the requirement for a complex series of compensatory mutations to maintain viral fitness. 65 CXCR4 co-receptor use by the virus changes over the course of infection from approximately 20% in early infection to approximately 50% in advanced disease with lower CD4+ cell count being a predictor of X4-using virus (reviewed in 60 ).66,67 This co-receptor switch, from predominant use of the CCR5 to the CXCR4 receptor correlates with disease progression i.e. increase in viral load and decline in CD4+ T-cell count. 68 However, whether the decline in CD4+ T-cells is the trigger for tropism switch or vice versa is unknown. In addition, Gonzalez 64 and colleagues have proposed that a decreasing levels of CXCL12 as resident DCs in lymph node are destroyed as disease advances also contribute to the X4 switch. Ribeiro and colleagues 69 have observed that R5 tropic HIV-1 preferentially enters memory CD4+ and X4-tropic HIV preferentially enters naïve CD4+ T-cells. The replication rate of memory cells early on HIV-infection is approximately 10 times the rate of naïve cells but as disease progression occurs the rates of replication become equal, implying that it is the change in rate of turnover of the naïve pool as disease progresses that drives this tropism switch. These authors also studied the effects of cART introduced before and after tropism switch. In both settings, virologically suppressive cART favoured the emergence of R5 tropic virus as the dominant quasi species, through a decrease in naïve cell turnover, which renders infection of X4-tropic virus so unproductive that reversion to R5 predominance was seen. The same was also true in the presence of CCR5-receptor blockers i.e. a paradoxical reversion to R5-tropic predominance. Pierdominici et al 70 showed that in patients treated and virologically suppressed on cART, levels of CCR5 expression on CD4+ and CD8+ T-cell subsets declined but the frequency of CXCR4 expression on naïve cells did not. In short, further data on the importance of tropism changes during long-term use of cART including cART which includes a CCR5-receptor blocker is needed with the caveat that evolution of the virus in the presence of virologically suppressive cART is unlikely.
Binding of HIV-1-Virus to CCR5
HIV-1 entry into host cells involves a complex interaction between HIV-envelope glycoprotein (Env) and cell surface receptors that ultimately result in a fusion reaction, not all of the steps are completely understood. Env comprises two subunits, the gp120 external subunit, which binds specific target cell receptors, and the gp41 trans-membrane subunit, which catalyzes the fusion reaction and anchors Env to the viral host-derived membrane. Steps in the sequence include binding of gp120 to CD4+ followed by a major conformational change that creates/opens up the CCR5 binding face. The sulphated N-terminus of the second extracellular loop of CCR5 and CXCR4 have been shown to physically associate with the CD4-activated bridging sheet and third variable (V3) loop of gp120 and by so doing allow the gp41 subunit to promote the fusion reaction via a series of complex conformational changes.71–78 Some of the difficulty in elucidating this process has been the hydrophobic nature of CCR5 meaning that the crystal structure of CCR5 when bound to CD4-gp120 is still not determined.
Delta-32 Homozygosity and ‘Resistance’ to HIV-1-Infection
In 1996, a deletion of 32 base pairs was identified in the CCR5 gene open reading frame resulting in a truncated dysfunctional protein not expressed on the cell surface. 79 This mutation on a single allele is common in Caucasian populations with a prevalence of 10% to 14%. Approximately 1% of Caucasians have 2 copies of this mutant gene, and the resultant phenotype is dramatically over-represented in populations of high-risk HIV seronegative persons who appear immunologically normal at least under most circumstances.79,80 Moreover, heterozygous individuals who have one CCR5 32 allele and one wild-type allele may be at marginally lower risk for HIV infection and appear to have an attenuated clinical course (in the absence of cART) with lower HIV viral loads and slower disease progression at least in the early stages of infection.81–83 Protection from HIV infection in those who are delta-32 homozygotes is not absolute; recent data has demonstrated that the mechanism of protection is by two different but linked pathways. First, the genetic loss of CCR5 expression on the cell surface makes the cell resistant to infection by R5 virus, in addition there is active downregulation of CXCR4 expression by the mutant CCR5δ32 protein making the cell relatively resistant to infection with X4 virus.84,85 In 2009, Huttner and colleagues 86 reported an HIV-infected patient who received a bone marrow allograft transplant from a donor homozygous for the CCR5δ32 mutation and subsequently does not have detectable HIV virus in plasma or tissues using current standard techniques; however, antibodies against HIV (gp41 and 120) remain detectable. This strategy is not one that could be broadly applied in clinical practice, but illustrates the proof of concept for CCR5 blockade as a strategic approach in the prevention of HIV transmission (discussed later).
Rationale for the Development of Inhibitors of Chemokine Receptor-5 (CCR5) as Anti-HIV Therapeutics
The rationale for the development of a new class of anti-HIV agents, CCR5-receptor antagonists, a host-directed therapy, as “antiviral agents” for HIV treatment is also drawn from the observations described above between chemokine receptors and resistance to infection and attenuated resultant disease. First, CCR5 is important for HIV entry and preventing this will have downstream effects on reducing HIV replication; second, as those with genetic deficiency of CCR5 appear “immunologically normal” (see later discussions), blockade of the CCR5 receptor as a therapeutic strategy will be immunologically benign. There are a number of strategies aimed at either blocking the CCR5 receptor directly i.e. the small molecule CCR5-receptor antagonists, which is the focus of this review, monoclonal antibodies such as PRO140 87 and drugs that down-regulate CCR5 receptor expression at the cell surface such as low dose rapamycin.88,89 In addition, there is some early data suggesting that a combination approach i.e. rapamycin to down-regulate the cell surface expression of CCR5 coupled with low doses of a small molecule CCR5-receptor blocker aplaviroc (no longer in development due to hepatotixicity) or its derivatives might be clinically applicable. 90 Clearly, since a host rather than viral factor is targeted, the mechanism of viral evasion of this approach has the potential to be quite different from the processes that generate viral resistance to other antiretrovirals.
Theoretical Risks of Blocking CCR5–- Infections and Oncogenic Potential
While the absence of CCR5 in people with the homozygous δ32 mutation appears benign under most circumstances, there are ongoing concerns in regards to the detrimental effects of blocking CCR5 in those whom CCR5 may play a pivotal role in the normal immune response to infections. These concerns extend to oncogenic viruses including Epstein-Barr virus (EBV) and the way immunological control might be altered in the setting of CCR5 receptor blockade. In the last five years, two infections, both encephalitides, West Nile virus (WNV) and Tick Borne encephalitis (TBE) have been associated with increased morbidity and mortality (7-fold increase with WNV) in patients homozygous for the delta 32 deletion.91–93 In mice models with CCR5 deficiency, central nervous system (CNS) infections with cryptococcus and Herpes simplex were problematical possibly due to a negative effect on leucocyte trafficking to the CNS.94–96 In addition, in CCR5 deficient mice, experimental T-cell mediated hepatitis was more severe. 97 Moreover, in humans homozygote and heterozygote for the delta 32 deletion, ischaemic-type biliary lesions post orthotopic liver transplantation (8 of 26 patients) were significantly more common than in those with wild-type CCR5 (14 of 120 patients) and moreover this had a negative impact on 5 year survival. 98 Another theoretical and as yet unstudied effect of CCR5-receptor blockade would be blunted vaccine responses.
Theoretical Benefits of Blocking CCR5–-Anti-Inflammatory Properties
Aside from these uncertainties in the context of CCR5-receptor blockers there may be some unexpected benefits if one extrapolates from observations in homozygotes for the delta 32 deletion. Overall, in some situations, there appear to be less harmful inflammation seen, examples of this include increased renal allograft survival; 99 lower incidence of RA, and in those with RA, fewer exacerbations; 100 less Kawasaki disease, 101 lower rates of severe coronary artery disease; 102 increased renal survival with IgA nephropathy 103 and in those with inflammatory bowel disease (IBD), less primary sclerosing cholangitis (PSC). 104 However, these data in the setting of IBD, conflict with those from another observational study which indicated that PSC was more severe in those heterozygotes for the delta 32 deletion. 105 Other observations include less inflammation and fibrosis in hepatitis C virus (HCV) and greater HCV clearance, 106 and lastly decreased cerebral malaria in mice. 107 To date no specific research has been conducted on the use of CCR5-receptor blockers as an adjunctive therapy in HIV-HCV or even HCV mono-infected patients.
Small Molecule Antagonists of Human Chemokine Receptor-5 (CCR5) in the HIV-Setting–-Focus on Maraviroc
Maraviroc–-mechanism of action
The small molecule chemokine receptor CCR5 antagonist, maraviroc, is licensed as a therapeutic agent in the treatment of HIV-1-infection in both the antiretroviral treatment experienced and naïve setting;108,109 other agents including vicriviroc are in Phase III development110,111 although the recent negative findings of the VICTOR-E3 and -E4 studies 112 have meant that the manufacturer of this agent will not submit a new drug application to the Food and Drug Administration for the use of vicriviroc in the treatment-experienced setting. 113 These agents are allosteric inhibitors that lock the CCR5 receptor into a conformation such that the receptor is not able to bind HIV envelope protein; the molecules also block, to various degrees, the signals induced by different receptor-binding chemokines. None of these agents is thought to promote receptor signaling or receptor internalization, therefore, these agents are not likely to promote inflammation–-as CCR5-receptor agonists might–-but, as the CCR5 receptor is maintained on the cell surface there is the potential for viral escape (see later discussions).
Tropism Determination, the Bête Noire of CCR5-Receptor Blockers
It was recognised early on in the development of the small molecule blockers of CCR5, that the degree to which the virus was utilising CCR5 to enter the cell, would impact on the efficacy of these agents in an individual. There are a number of phenotypic assays for tropism; those developed by Monogram Biosciences are the most widely utilised. 114 The Trofile test 114 reports tropism following infection of reporter cell lines by pseudotyped viruses (which lacks a functional gp-160 gene but containing a lucifersae reporter gene) carrying patient derived Env. These pseudo-typed viruses are separately incubated with CCR5 and CXCR4 expressing CD4+ T-cells, and luciferase activity in cell lysate is measured 72 hours later. 115 This allows the tropism of the patient's virus to be determined as R5-using, X4-using or dual tropic. It is a prerequisite of the licensure for maraviroc that tropism of a patient's virus has been determined as R5 using a licensed tropism assay prior to the drug being prescribed; in general the most widely used assay is the second generation and more sensitive monogram Trofile® ES assay or ESTA. 114 Compared to the original tropism assay utilised in the licensing studies for maraviroc, this Trofile test can detect mixtures of R5/X4 of 0.3% and is about 30-fold more sensitive than its predecessor. However, there are also other tropism assays in development such as the SensiTrop II, by Pathways Diagnostics116,117 and more recently, the Toulouse Tropism Test (TTT) 118 (see discussions below).
A requirement for a phenotypic test prior to utilisation of this class of agents represents a major challenge in clinical practice. First the test is not locally available in many countries, necessitating samples be sent to the US for processing. Currently the manufacturer of maraviroc facilitates access to the phenotypic test in some countries, but in time, this not insubstantial expense will have to be shouldered by public health authorities in countries which approve the use of the drug. In wealthier countries, this may well be possible, but in developing nations, the cost of the tropism assay coupled with the expense of the drug will be prohibitive as the test will need to be available prior to initial use of the drug and during viral rebound. Moreover, detectable plasma viraemia is required to reliably conduct a phenotype, with the best chance of successful tropism determination if plasma HIV RNA is >1000 copies/mL. The latter has seriously hampered switch studies or indeed intensification studies where tropism cannot be determined when plasma virus is suppressed (usually <50 copies/mL).
Over the last five years this has been an area of intensive research as is clear that high throughput genotypic testing is needed as a reliable alternative to phenotypically derived tropism. Different sets of rules have been published based on the amino acid sequence of the Env-V3 region of HIV-gp120, known to be the major determinant of co-receptor usage119,120 and among these, the most widely used is the 11/25 rule. Raymond and colleagues 121 compared their genotypic classification using the 11/25 rule to predict CXCR4 usage (an R or K at position 11 of V3 and/or K at position 25; R at position 25 of V3), combined 11/25 rule and net charges rule (an R or K at position 11 of V3 and/or K at position 25; R at position 25 of V3 and a net charge (positively charged minus negatively charged amino acids) of ≥+5) and two different bioinformatics driven prediction systems, geno2pheno 122 and WebPSSM. 123 Overall, for detecting CXCR4, sensitivity and specificity of the genotype was 77% and 96% (combined 11/25 rule and net charges rule), 88% and 87% (Geno2Pheno) and 77% and 94% (WebPSSM tool) respectively. 121 Other researchers have subsequently explored whether the predictive value of genotypic tests can be enhanced by including clinical information in the prediction tool as well exploring the application of these rules to non-Clade B viruses. For the most part the genotypic testing appear to perform as well in Clades A and C as for Clade B viruses.124–126
More recently, a number of investigators have presented data on the determination of HIV-1 co-receptor tropism using proviral DNA derived from peripheral blood mononuclear cells (PBMC) and on deep sequencing of the V3 loop. The real appeal of the former is that once validated in larger numbers of patients, this methodology will facilitate the use of CCR5-receptor blockers in switch and/or intensification studies in aviraemic patients. Toma and colleagues 127 sequenced proviral-derived Env sequences which were subsequently processed through the Trofile assay. PBMC and plasma tropism determinations were concordant (8 R5, 2 DM) and overall, there was good homology between the PBMC proviral DNA-derived and plasma derived viruses in the 10 aviraemic patients. Swenson and colleagues 128 investigated a different approach. Here they compared virological responders vs. non responders using three different methods to determine tropism i.e. the original Trofile test, the Enhanced Sensitivity Trofile Assay (ESTA) and population-based V3 deep sequencing (samples were classified as X4 if ≥2% of the virus population scored ≤3.5 using the geno2pheno algorithm) from a number of maraviroc studies. The ESTA and deep sequencing of the V3-loop were superior in predicting virological response in the naïve and treatment-experienced setting compared to the original Trofile test and deep sequencing appeared to have higher specificity than the ESTA.
Raymond and colleagues 118 have developed the TTT test as an alternate to the Trofile assays. The TTT utilises cell associated HIV DNA to determine tropism and successfully determined tropism in 97% of samples, both from patients with low HIV RNA and with different HIV Clade. TTT was able to detect X4 mixtures of 0.5%.
Despite the advances in determining tropism through genotypic and phenotypic testing, many challenges remain in addition to those described above. Clearly there are limitations to the algorithyms of genotypic prediction that only look at the V3 loop, as there may be down-stream changes that impact on phenotypic tropism. Next, while we have tropism assays that are now very sensitive, we do not yet know where to set the bar for dual tropic mixtures in terms of clinical threshold. The latter may vary depending on the circumstance under which the CCR5-receptor blocker is being used (e.g. ART-naïve, first failure, “multi-drug resistance”). Last, another problem in clinical practice is patients who have an “unreportable” Trofile test, Genebat and colleagues 129 have described the utilization of short-course exposure (8 days) to MVC as a surrogate for tropism testing. They defined “response” as plasma viral load below 40 copies/mL or >1 log10 copies/mL reduction by Day 8; and found that this MVC clinical test (MCT) showed concordance with the Trofile in 93.5% of patients.
Maraviroc in the Antiretroviral Treatment Experienced Setting
Maraviroc, a small molecule antagonist of chemokine receptor-5 (CCR5), received accelerated approval in 2007 as an anti-HIV agent for use in patients with multi-drug resistant (MDR) R5-tropic HIV based on the results of the MOTIVATE 1 and 2 studies.40,130 In MOTIVATE 1 and 2, 1049 patients were randomized to receive either once or twice daily maraviroc (MVC) with optimized background regimen (OBR) vs. OBR alone. Mean viral load reductions (log10 copies/mL) at week 48 were -1.66 and -1.72 with the once daily dose of MVC in MOTIVATE 1 and 2 respectively; -1.82 and -1.87 in MOTIVATE 1 and 2 respectively for the bid MVC dosing groups, versus -0.8 and -0.76 in the control (non-MVC) arms of MOTIVATE 1 and 2 respectively. The respective response rates (for viral load suppression to <50 copies/mL) were 42% and 45% in the once daily MVC dose in MOTIVATE 1 and 2 respectively; 47% and 45% for the twice daily MVC arm in MOTIVATE 1 and 2 respectively and 16% and 18% in MOTIVATE 1 and 2 respectively for the OBR only arm (P = <0.001 for the pairwise comparisons). Moreover, there were greater increases in CD4+ T-cells between baseline and week 48 in the MOTIVATE 1 (+113 vs. +122 vs. +54 cells/uL in the once daily MVC vs. twice daily MVC vs. OBR only arms respectively) and 2 (+122 vs. +128 vs. +69 cells/uL in the once daily MVC vs. twice daily MVC vs. OBR only arms respectively) studies compared to the OBR only group (P = <0.001 for pairwise comparisons). 130 Importantly the CD4+ T-cell gain in the MVC arms was still significantly greater (P = 0.05) compared to the non-MVC arms in those with viral load suppression, suggesting an effect on immune restoration over and above the antiviral effects of the MVC-containing regimens. 131
Even in patients who never achieved full viral load suppression, there were greater gains in CD4+ T-cell counts in the MVC arms compared to the non-MVC arms, and these gains were preserved through to week 48 (P = 0.0013). In addition, early gains in CD8+ T-cells were observed in all three arms, but these were most pronounced in the MVC arms (median gain over 48 weeks of +152 vs. +141 vs. +9 cells/uL in the once daily MVC vs. twice daily MVC vs. OBR only arms respectively); while these levels declined after week 24, the levels had not normalized in the MVC arms by week 48 due to the initial large gains. It is unclear whether the significant increase in time to Category C events in the MVC compared to the control arm is related to improved virological suppression, additional gain in CD4+ T-cells or both.
We have explored this phenomenon in a smaller Phase 1 study. 132 Here we exposed patients to monotherapy with an experimental CCR5-receptor blocker (SCH532706) over 10 days. We found a profound increase in circulating CCR5+CD8+ T-cells during the period of dosing, with changes occurring very early and preceding any change in HIV viraemia. This suggested an effect on CD8+ trafficking during CCR5 receptor blockade.
There have been concerns that the use of CCR5-receptor blockers might be associated with negative immunological consequences i.e. through the exertion of selection pressure on the virus to undergo tropism switch and this might result in disease progression. However, in a subgroup analysis of MOTIVATE 1 and 2, there was no evidence of CD4+ T-cell decline following MVC failure secondary to the emergence of X4 virus. 133 Moreover, in an exploratory study, in which patients with dual/mixed tropic or X4 virus 134 were treated with MVC and OBR vs. OBR alone, while viral load reductions (log10 copies/mL) over 24 weeks were equivalent i.e. -0.97 in the OBR arm vs. -0.91 (once daily MVC+OBR) vs. -1.20 (twice daily dosing MVC arm) the CD4+ T-cell response in the MVC arms was significantly better than the OBR only arm i.e. +60 and +62 cells/uL in the once and twice daily MVC arms respectively vs. +36 cells/uL in the OBR arm. The incidence of serious adverse events was similar between all arms.
Maraviroc in the Antiretroviral Naïve Setting
The MERIT study (
Maraviroc Dosing, Drug-Interactions and Special Populations
Dosing and potential drug interactions
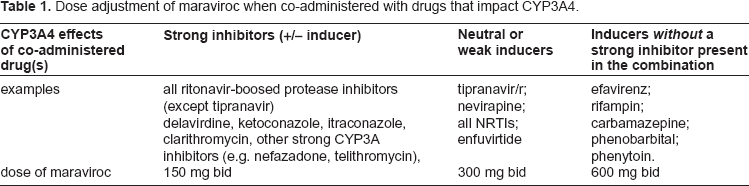
Early dose ranging studies the drug and the Phase III studies provided information as to the correct dosing of maraviroc 40 at least when partnered with an NRTI-backbone in the naïve setting and when coupled with various OBR in the treatment-experienced setting. The drug is available as both a 150 mg and 300 mg tablet and can be dosed with/without food, with no dose adjustment based on ethnicity or gender. The usual dose of maraviroc is 300 mg bid, but as the drug is a substrate of CYP3A and Pgp its pharmacokinetics are affected by the co-administration of inhibitors and inducers of these pathways. The product label advises that when maraviroc is co-administered with the following, dose adjustment is required (Table 1).
Dose adjustment of maraviroc when co-administered with drugs that impact CYP3A4.
Pregnancy
Maraviroc appears safe in animal studies with rats and rabbits exposed to levels 5–20 fold higher than human exposures showing no teratogenicity or post-natal developmental abnormality (including fertility and reproductive potential) in offspring. However, despite this reassuring data from non-human species, maraviroc remains a Category B drug because of the paucity of safety data in pregnant women exposed to the drug.
Renal Impairment
Approximately 25% of total clearance is renal, and hence in those with renal impairment, it seems likely that levels will be increased especially when CYP3A inhibitors are co-administered. The product information suggests those with a creatinine clearance of <50 mL/min receiving maraviroc co-administered with a CYP3A inhibitor may be at increased risk of adverse effects such as dizziness and postural hypotension secondary to increased levels of maraviroc. No dose reduction is recommended in this setting, but closer clinical monitoring is advised.
Maraviroc Resistance Pathways
It has been recognised over the past five years, that there are two main mechanisms of resistance to the small molecule CCR5-receptor antagonists. Escape can occur both through the emergence of X4-tropic virus or the continued use of the CCR5-receptor bound to the small molecule blocker by R5-virus (reviewed in140,141). The latter appears to occur through accumulation of between 2–4 sequence changes in the gp120 V3 region and can exist de novo in patients who have never been exposed to CCR5-receptor blockers.142,143 These maraviroc resistant CCR5-tropic viruses give dose response curves that are characterized by plateaus in maximum percentage inhibition (MPI) below 100%. However, mutations downstream to the V3 loop can also result in this type of resistance emerging. Moreover, the latter does not appear to have a fitness cost to the virus.144,145 The relative contribution to these different resistance pathways in viral failure in vivo, has been demonstrated in the maraviroc licensing studies. In the MERIT study 109 for 29 maraviroc-treated and 13 efavirenz-treated patients who experienced virologic failure by the TLOVR (time-to-loss of virological control) algorithm (<50 copies/mL threshold), 31% of maraviroc failure was due to the emergence of CXCR4-using virus (9/29); 14% (4/29) due to selection of CCR5-tropic virus with resistance to maraviroc; 17% (5/29) failed with only lamivudine-resistant virus and 38% (11/29) failed with no discernible resistance patients, probably due to periods of poor adherence. Of the 13 efavirenz failures, 69% had NNRTI resistance; of these 9 patients, 4 had resistance to lamivudine and 1 had resistance to zidovudine. Moreover, 5 patients in the efavirenz group discontinuing ART because of adverse events, developed new NNRTI resistance during follow up. This was probably due to the long half-life of efavirenz without other antiretrovirals given to cover this “NNRTI tail”; this did not occur with maraviroc discontinuations.
Maraviroc Side-effect Profile
The most common adverse events reported with maraviroc given twice-daily, and occurring at rates higher than seen in the placebo arm for which causality may or may not exist include: cough, pyrexia, upper respiratory tract infections, rash, musculoskeletal symptoms, abdominal pain and dizziness. Diarrhoea, oedema, influenza, oesophageal candidiasis, sleep disorders, rhinitis, parasomnias and urinary abnormalities were also seen at higher rates with the once daily dosing compared to either the bid dosing arm or placebo.
Overall, in the MOTIVATE-1 and -2 studies, 130 adverse events at the week 24 interim analysis were similar to those in the comparator (i.e. optimised background therapy without maraviroc) arm. Five percent or fewer study participants in both arms discontinued treatment because of adverse events. Side-effects reported with the use of maraviroc and of particular importance 40 are summarised below.
Hepatotoxicity
The liver was considered a potential target organ for toxicity with maraviroc as data arising from the preclinical rat studies showed toxicity at supra-therapeutic levels; moreover, another small molecule blocker of CCR5, aplaviroc was discontinued, due to severe idiosyncratic hepatotoxicity. However, overall grade 3–4 hepatotoxicity was low in all of the maraviroc licensing studies109,130 and comparable to the placebo arms. That being said, there were two cases of severe hepatotoxicity that should be noted. The first occurred in a healthy volunteer, with maraviroc-induced hepatoxicity with allergic features. As a consequence, the product information suggests urgent assessment +/-product discontinuation if any sign of a systemic allergic reaction appears following maraviroc commencement. The second, occurred in one of the naïve studies. Here, a maraviroc recipient developed acute liver failure necessitating a liver transplant. This event was complicated by the co-administration of isoniazid (INH) and cotrimoxazole and both drugs were started shortly before the commencement of cART. Moreover the patient was subsequently found to be a slow acetylator for the NAT2 alleles and carried the CYP21E genotype, increasing further the risk of INH hepatotoxicity. 40
Maraviroc has not been specifically studied in patients with hepatitis B and or C infection or in those with pre-existing liver disease. In the MOTIVATE studies 130 and MERIT studies, 109 although co-infection rates with hepatitis C were higher than with Hepatitis B, both were still very low (in MERIT, (7.8% in maraviroc and 6.6% in the efavirenz arms), there was no excess of hepatotoxicity seen in those co-infected.
In October 2009, the Food and Drug Administration 139 approved the use of maraviroc in an ART-naïve setting. The rationale for this was for two main reasons. First, the equivalent performance of the drug (vs.efavirenz) when the ESTA was used to identify those with R5-using virus. Second, increased tolerability of the drug compared to efavirenz in the post hoc analysis of MERIT 109 as exemplified by the low adverse event rates, lipid neutrality and lower rates of discontinuation.
Despite this approval, the DHHS panel 4 have determined that there is still insufficient evidence to recommend maraviroc as first line therapy for antiretroviral naïve patients. At least one naïve study utilising maraviroc partnered with a ritonavir-boosted protease inhibitor as part of an NRTI-sparing regimen is ongoing; data is expected in 2011.
Infections and Malignancies in patients Treated with CCR5-Receptor Blockers
There are ongoing concerns as to whether the CCR5-receptor blockers might be associated with an increased risk of and/or impaired handling of some infections in which CCR5 may play a pivotal role for the trafficking of effector cells. As discussed earlier in this review, these theoretical concerns extend to oncogenic viruses including EBV and the way immunological control might be altered in the setting of CCR5 receptor blockade. However, this anxiety does not appear to have been borne out in clinical practice at least in the short term, although there were some early concerns regarding malignancy (4 cases of lymphoma) in a Phase II vicriviroc study, ACTG5211. 146 The authors were able to demonstrate that there was no up regulation of EBV replication (as measured by viral load) in recipients of vicriviroc (n = 116), and only one of four lymphoma cases was EBV positive. In the MERIT study, 109 time to category C events was longer in those receiving maraviroc compared to efavirenz (2.5% compared with 3.3%), with fewer AIDS defining and non-AIDS defining malignancies including lymphoma (1.4% compared with 3.3% maligancies). Overall, in regards to other infections, an increased incidence of upper respiratory tract infection, candida infection and Herpes infections in those receiving maraviroc vs. placebo in the treatment-experienced studies was observed; 40 however, the use of maraviroc was associated with a decreased incidence of pneumonia.
In summary, while there is no worrisome signal in regards to infections and malignancy, the data derived from clinical studies and the post-marketing experience, is small and of relatively short duration. The authors endorse the view that in the absence of long term data on the use of this class of agents in HIV-infection, continued vigilance and research is warranted.
Maraviroc as an Immunomodulating Agent
The data presented earlier in this review and the results of a meta-analysis by Wilkin and colleagues 147 showing increased CD4+ T-cell increase with CCR5-receptor blockers has fuelled interest in the use of these agents as immunomodulating drugs. The mechanism of these CD4+ T-cell increases is unclear as is the clinical significance. One proposed mechanism is that these agents may have a role in reducing immune activation. 148 It is currently unknown whether the putative “antiinflammatory” effects of CCR5 blockade contribute to the “improvement” in CD4+ T-cell count over and above CD4+ T-cell immune restoration secondary to their antiviral effects. As discussed previously, there is increasing recognition of the role of chronic immune activation in the pathogenesis of HIV infection. 27 In part this may be mediated by ongoing CCR5 signalling through persistent antigen, either as consequence of ongoing HIV replication or in response to other latent viruses/bacteria which might include Herpes simplex, cytomegalovirus, and leakage of gut pathogens. However, at the present time, it is unclear what the relative contribution (if any) of the anti-inflammatory activity of CCR5-receptor blockers is in regard to immune restoration in the HIV setting. It is noteworthy that until very recently, maraviroc was being developed as an anti-inflammatory agent in the setting of rheumatoid arthritis.149,150 However, disappointingly, this phase II study was discontinued due to lack of efficacy. 151 However, despite the lack of convincing data for maraviroc as an anti-inflammatory agent, at least in the setting of RA, there is still interest in intensifying cART with a CCR5-receptor blocker in those with poor CD4+ immune restoration despite full viral suppression. 152 The interest is twofold, first to enhance HIV viral suppression in compartments such as central nervous system (CNS) and genital tract and second, to harness the putative role of maraviroc as an anti-inflammatory agent. Early data presented by Evering and colleagues 30 suggested no effect of maraviroc intenstification on immune activation or virological parameters in the GALT. Wilkin and colleagues 31 in a pilot study of MVC intensification, showed modest decreases in immune activation as evidenced by reduced percentage CD38+ and HLA-DR+/CD38+ expression and reduction in apoptosis without any change in peripheral CD4+ or CD8+ T-cell levels.
Maraviroc as Part of Pre-exposure Prophylaxis (prEP)
Extending the scope of pre-exposure prophylaxis from the prevention of mother-to-child transmission to prophylaxing HIV negative individuals using oral or topical formulations of antiretroviral agents to prevent HIV acquisition are strategies being researched. Candidate orally administered antiretrovirals include tenofovir, tenofovir with emtricitabine or maraviroc (reviewed in 153 ). MVC seems an ideal candidate as the majority of transmitted HIV is R5-using (reviewed in 154 ); moreover the drug is concentrated in the genital tract 155 and has a favourable side-effect profile. 40 The other strategy is the use of topical formulations of antiretrovirals and those using tenofovir applied as a vaginal gel are most advanced in terms of development. 156
CCR5-Receptor Blockers, Future Research Directions
The approval of maraviroc in the treatment naïve setting based upon the reanalysis of the MERIT study 109 has led to renewed efforts to find a niche role for this agent. Some of the areas of research, not unique to maraviroc per se, but of importance in clinical practice are listed below:
As part of an NRTI sparing regimen. There are increasing concerns regarding the toxicity profile of NRTI,6,7,157 and as a consequence, there is interest in exploring the use of NRTI-sparing regimens. Moreover, as the terminal half-life of MVC is 14–18 hours, this agent has the potential for once-daily dosing (although qd dosing failed in MERIT possibly because of incorrect tropism assignment. 109 Once-daily MVC dosing (150 mg) with ritonavir-boosted atazanavir is being explored in a randomised trial in antiretroviral naïve patients. 158
In primary HIV infection, to reduce the early loss of CD4+ T-cells from the GALT 159 and to enhance restoration of GALT in chronic infection; 160
As an “intensifying agent” to reduce the HIV reservoir;161,162
As part of a “neuro-effective” antiretroviral regimen in patients with existing HIV associated neurocognitive disease (HAND) or indeed to prevent HAND. The rationale for consideration of CCR5-receptor blockers in this setting is two-fold. First, CNS HIV strains appear to be largely R5-using even when plasma viruses are dual-tropic/X4163–165 and second CNS penetrance is reasonably high–- many believe the latter might be important in the treatment and/or prevention of HAND. Although maraviroc (and vicriviroc) only attains central nervous system (CNS) levels which are 10% of those in plasma, at least in the rat model (n = 4), these levels will still be therapeutically relevant. Moreover the CNS effectiveness penetrative score (CPE) for both of these small molecule blockers of the CCR5 receptor, based upon pharmacokinetic (PK) characteristics including plasma protein binding, Cmin and IC50 suggest that both drugs have high CPE scores;166,167 (Scott Letendre personal communication) Ongoing clinical trials exploring the latter are currently in progress. 168
Conclusion
Maraviroc is the first-in-class licensed small molecule antagonist of the CCR5 receptor and as such is a welcome addition to the armamentarium of drugs for the treatment of HIV-infected adults. This host-directed therapy blocks HIV viral entry into cells expressing CCR5 and by so doing functions as an antiviral agent. The utility of this class as immunotherapeutic agents is being explored particularly in the setting of those with an immunodiscordant response to virologically-suppressive cART.
In clinical practice, this class of agent presents a particular challenge because of the requirement to assess virus tropism prior to use. This phenotypic test is costly, and access to the test limited; in addition there is an added challenge of assessing tropism in aviraemic individuals and currently, sequencing of proviral DNA for subsequent assessment of tropism remains experimental. If the issues of tropism testing are overcome, it makes the most sense to utilise this drug early on in the course of HIV infection when virus is more likely to be R5. MVC-based regimens are approved for use and likely will be recommended in the ART-guidelines for ART-naïve patients. If this eventuates, clinicians and their patients can choose between raltegravir or PI/r or NNRTI or CCR5-receptor blocker based regimens as first-line therapy. Faced, with such choices, the relative merits of each combination will need to be weighed-up carefully and plans for switching for toxicity and first failure need to be developed, preferably with robust data to support the strategy.
Last, despite the lack of any particular safety signal in the maraviroc licensing studies, there is still concern regarding the potential long-term adverse effects of blocking a key cellular trafficking receptor in hosts who do not have genetic absence of this pathway.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.