
Abstract
Sustained inhibition of HIV-1, the goal of antiretroviral therapy, is often impeded by the emergence of viral drug resistance. For patients infected with HIV-1 resistant to conventional drugs from the viral reverse transcriptase and protease inhibitor classes, the recently approved entry and integration inhibitors effectively suppress HIV-1 and offer additional therapeutic options. Entry inhibitors are particularly attractive because, unlike conventional antiretrovirals, they target HIV-1 extracellularly, thereby sparing cells from both viral- and drug-induced toxicities. The fusion inhibitor enfuvirtide and the CCR5 antagonist maraviroc are the first entry inhibitors licensed for patients with drug-resistant HIV-1, with maraviroc restricted to those infected with CCR5-tropic HIV-1 (R5 HIV-1) only. Vicriviroc (another CCR5 antagonist) is in Phase III clinical trials, whereas the CCR5 antibodies PRO 140 and HGS 004 are in early stages of clinical development. Potent antiviral synergy between maraviroc and CCR5 antibodies, coupled with distinct patterns of resistance, suggest their combinations might be particularly effective in patients. In addition, given that oral administration of maraviroc achieves high drug levels in cervicovaginal fluid, combinations of maraviroc and other CCR5 inhibitors could be effective in preventing HIV-1 transmission. Moreover, since CCR5 antagonists prevent rejection of transplanted organs, maraviroc could both suppress HIV-1 and prolong organ survival for the growing number of HIV-1 patients with kidney or liver failure necessitating organ transplantation. Thus, maraviroc offers an important treatment option for patients with drug-resistant R5 HIV-1, who presently account for >50% of drug-resistance cases.
Introduction
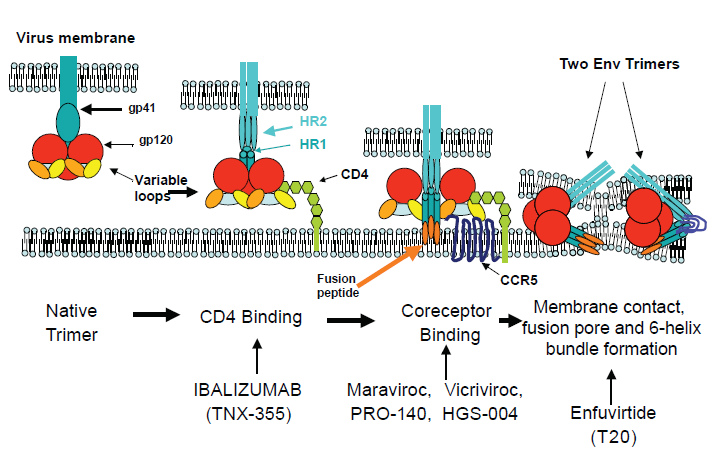
Highly Active Antiretroviral Therapy (HAART), through the combination of nucleoside analog reverse transcriptase inhibitors (NRTIs), non-NRTIs (NNRTIs) and protease inhibitors (PIs), has significantly changed the progression and outcome of infection with HIV-1. 1 However, high pill burdens, inconvenient dosing, and long-term toxicities contribute to poor compliance and emergence of drug-resistant virus in many patients.2,3 For those patients in whom resistant virus develops, treatment options become limited and more complicated regimens are necessary to prevent further disease progression. Fortunately, the elucidation of HIV-1 entry steps has offered new opportunities for therapeutic intervention 4 (Fig. 1). Two entry inhibitors are currently licensed, the fusion inhibitor enfuvirtide (T-20) 5 and the small-molecule CCR5 antagonist maraviroc. 6 Vicriviroc, another CCR5 antagonist, is in advanced clinical development. 7 The CCR5 antibodies PRO 140 8 and HGS004, 9 and the CD4 antibody ibalizumab 10 are in early clinical development. Entry inhibitors, as well as integrase inhibitors and newer NNRTIs and PIs, have demonstrated potent HIV-1 inhibition in treatment-experienced patients, with significantly improved suppression when at least two active drugs were used.11–13 Accordingly, current treatment guidelines recommend the use of at least two, and preferably three, fully active agents in a new regimen in patients with evidence of HIV-1 resistance. 14 This review will focus on maraviroc (Selzentry, Pfizer Inc), the first licensed CCR5 antagonist for patients with drug-resistant CCR5-tropic HIV-1 infection.
Maraviroc: Mechanism of Action and Factors Impacting Antiviral Activity
Maravirocis aspirodiketopiperazinethattargetsCCR5, a main coreceptor for HIV-1. 15 The identification of CCR5 as a viral coreceptor16–19 was prompted by the discovery of HIV-1 inhibition by the CCR5 ligands β-chemokines (MIP-1α [CCL-1], MIP-1β [CCL-2] and RANTES [CCL-3]). 20 CCR5, which belongs to the G-protein-coupled receptor (GPCR) family and is expressed on activated T lymphocytes, macrophages and dendritic cells, regulates cell trafficking to inflamed tissues. 21 CCR5-tropic HIV-1 strains (referred to as R5 HIV-1) are the most persistent and predominantly transmitted ones.22,23 HIV-1 strains that use the alternative chemokine receptor CXCR4 (X4 HIV-1) or both CCR5 and CXCR4 (R5X4 dual-tropic HIV-1) emerge in approximately 50% of patients and are generally associated with a more rapid loss of CD4+ lymphocytes and faster progression to AIDS.24,25 However, transition to CXCR4-using HIV-1 strains is not required for the development of AIDS since CD4 depletion and disease progression does occur in patients carrying R5 HIV-1 strains only. 26 Several polymorphisms that impact CCR5 expression have been described. A mutation in the CCR5 open reading frame leads to premature truncation and consequently a 32-bp deletion in the CCR5 protein (CCR5δ32). Individuals homozygous for the δ32 mutation (~1% in the Caucasian population) are generally resistant to infection with HIV-1, while heterozygous individuals can acquire infection but progress to AIDS more slowly than wild-type individuals.27,28
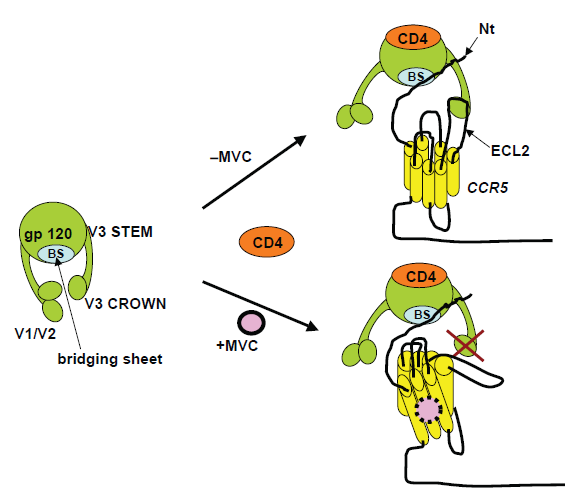
To infect a host cell, the envelope (Env) proteins on the surface of HIV-1 first bind to cellular CD4 receptors. Env bound to CD4 receptors then interacts with the coreceptor (CCR5 or CXCR4) to trigger the fusion of viral and cellular membranes leading to viral entry into cells. 4 In R5 HIV-1 infection, following CD4 engagement, the CCR5 N-terminus binds to the bridging sheet (formed between the C1, C2 and C4 domains) and basal V3 regions of gp120 whereas the CCR5 extracellular loops bind to the tip of V3. 29 Small-molecule CCR5 antagonists, such as maraviroc, bind to a hydrophobic cavity formed by the first, second, third and seventh transmembrane domain helices of CCR5 (Fig. 2). Within this cavity, maraviroc interacts with amino acids Trp86, Glu283, Tyr108, Tyr251 and Ile198, inducing conformational changes in the extracellular loops that are not recognized by the HIV-1 V3 region.30–34 Thus, CCR5 antagonists act as allosteric, non-competitive inhibitors. Unlike the natural β-chemokine CCR5 ligands, which induce transduction signaling and coreceptor internalization,35,36 antagonist-bound CCR5 does not signal and remains on the cell surface. Several factors, including densities of CCR5 and CD4 on the cell surface as well as affinity of HIV-1 Env for CCR5 can impact the efficiency of viral binding, entry, and infection.
Effect of CCR5 Density on Antiviral Activity of CCR5 Antagonists
Early studies with cell lines demonstrated that CCR5 density (molecules/cell) can limit HIV-1 entry, with a threshold below ~2 × 10 3 CCR5 molecules/CD4+ cell resulting in inefficient infection. 37 In addition, coreceptor density on cell lines influences the kinetics of fusion and therefore susceptibility of R5 HIV-1 to the fusion inhibitor enfuvirtide.38–40 However, one caveat of studies using enfuvirtide in cell lines is that they may not accurately reflect the situation in primary CD4+ T cells. In this regard, CCR5 levels among cell lines may vary by several orders of magnitude (from <7 × 10 2 to >10 5 molecules/cell), whereas CD4+ T cells from normal individuals vary only by ~5-fold (2-10 × 10 3 CCR5 molecules/cell).21,37,41,42 In an effort to evaluate the impact of CCR5 expression on HIV-1 entry inhibitor activity, we showed a positive correlation between CCR5 density on primary CD4+ T cells and decreased sensitivity of R5 HIV-1 strains to enfuvirtide. 43 In addition, we demonstrated that inhibition of CCR5 expression by the immunomodulatory drug rapamycin synergistically enhanced enfuvirtide activity against R5 HIV-1. 44
It is reasonable to think that the activity of CCR5 antagonists will depend on the amount of CCR5 available on the cell surface. Indeed, Reeves et al have shown that lower concentrations of the CCR5 antagonist TAK-779 were required to inhibit the infection of cell lines engineered to express low CCR5 levels compared to cells expressing high CCR5 levels. 39 Willey et al have reported similar results using the cell line U87 and several CCR5 inhibitors. 45 Moreover, Platt et al demonstrated that TAK-779 was ~ 15-fold more potent against infection of HeLa-CD4 cells expressing low levels of CCR5 (~2 × 10 3 CCR5 molecules/cell) than of cells with higher CCR5 levels (~2 × 10 4 CCR5 molecules/cell). 38 However, as indicated above, studies of CCR5 antagonists conducted in cell lines may not reflect the situation in primary CD4+ T cells. In addition to differences in coreceptor expression levels between cell lines and primary cells, it is possible that differences may exist in processing or post-translational modification. For example, differences at the processing/post-translational level could result in altered antagonist affinity. Our results using vicriviroc demonstrated that CCR5 levels on donor lymphocytes correlated with its antiviral activity against R5 HIV-1. Moreover, we showed that reduction of CCR5 expression by rapamycin enhanced the antiviral activities of the CCR5 antagonists TAK-779 46 and vicriviroc 47 in primary CD4+ T cells.
Interdependence of CCR5 and CD4 Levels on HIV-1 Entry
Studies on cell lines have demonstrated that densities of CD4 and CCR5 required for R5 HIV-1 infection are interdependent. 37 Cell lines with a high CD4 density (4.5 × 10 5 receptors/cell) require low levels of CCR5 (~2 × 10 3 receptors/cell) for infection, whereas cells with low CD4 (<10 4 receptors/cell) require a higher CCR5 level (1-2 × 10 4 receptors/cell). Primary CD4+ lymphocytes express ~2-10 × 10 3 CCR5 receptors/cell and ~2 × 10 4 -1 × 10 5 CD4 receptors/cell. 21 Accordingly, small changes on levels of CCR5, but not CD4, on donor lymphocytes may be expected to impact R5 HIV-1 infection. In support of this hypothesis, we have reported that changes on CCR5 density, but not CD4 density, on donor lymphocytes impact infectivity by R5 HIV-1. 43
Reciprocal Modulation of CCR5 and β-Chemokine Levels
Expression of the CCR5 receptor on lymphocytes is transcriptionally controlled by cellular activation and requires interleukin-2 signaling for continuous expression. 48 Transcription is initiated at multiple sites in exons 1 or 2 and alternative promoter usage gives rise to different transcripts. 49 Early studies in transformed cell lines showed that CCR5 transcription is mainly driven by promoter 1 (Pr 1), leading to the assumption that Pr 1 governs the expression of CCR5 on primary cells.49,50 However, a study by Mummidi et al has demonstrated that, unlike in cell lines, CCR5 transcription in primary lymphocytes is mostly driven by Pr 2. 51 As pointed out earlier on this review, these findings underscore the importance of using primary lymphocytes in studies of CCR5 expression and HIV-1 entry inhibition. This study also showed that synthesis of the CCR5 mRNA isoforms CCR5A and CCR5B is associated with the level of CCR5 surface expression on lymphocytes. Moreover, the transcription factors Oct-2 was shown to enhance the synthesis of these isoforms and CCR5 surface expression, a finding that extended early observations by Moriuchi et al. 52
The levels of extracellular β-chemokines and CCR5 receptors are reciprocally modulated via internalization of chemokine-bound CCR5 receptors. Increased production of CCL3L1 (MIP-1αP) by individuals carrying a duplication in the CCL3L1 gene results in greater internalization of CCR5 and therefore fewer CCR5 receptors on the cell surface. 53 Similarly, individuals who are homozygous for the δ32 deletion in the CCR5 gene and as a result lack CCR5 protein expression, show high levels of β-chemokines in culture supernatants due to their inability to induce internalization.54,55
Impact of HIV-1 Diversity on Antiviral Activity of CCR5 Antagonists
Targeting components of the HIV-1 Env proteins (gp120 and gp41) with an entry inhibitor faces the challenge of genetic variation. The Env gene is the most variable HIV-1 gene, with up to 30% diversity among clades, 20% diversity within a clade, and up to 10% diversity within an individual. 56 Within gp120, the bridging sheet and V3 regions participate in coreceptor binding. 57 Although the exact epitope on gp120 that interacts with the coreceptor is not known, it is likely to differ among HIV-1 strains. However, currently available data indicate that most viruses, from same or different subtypes, are similarly inhibited by maraviroc (IC90 of 2 nM; 95% CI of 1.8 to 2.4 nM). 15 In agreement, all patients from a maraviroc phase IIa trial had viral load reductions of >1 log10. 58 One exception to the broad antiviral activity of maraviroc is Subtype G HIV-1, which is less sensitive to maraviroc and to vicriviroc, at least in vitro.15,59
Maraviroc Pharmacokinetics
Maraviroc is orally administered, 300 mg twice daily without regard to food. The drug is rapidly absorbed, with peak drug concentrations between 0.5 and 4 h after oral dosing. 60 At the licensed dose of 300 mg, maraviroc has a bioavailability of 33% and a terminal t1/2 to steady state of 14-18 h, with steady state reached within 7 days. 61 Maraviroc is substrate for CYP3A4 and p-glycoprotein, and thus, doses need to be adjusted when coadministered with inhibitors or inducers of these pathways as follows: 150 mg twice daily in the presence of CYP3A4 inhibitors (PIs, delavirdine, ketoconazole, itraconazole, clarithromycin), and 600 mg twice daily in the presence of CYP3A4 inducers (efavirenz, rifampicin, carbamazepine, phenobarbital and phenytoin).62,63 The standard dose of 300 mg twice daily can be used in the presence of tipranavir/ritonavir, nevirapine, NRTIs and enfuvirtide.
Maraviroc has poor penetration into the CNS of rats, suggesting it has limited antiviral activity in the brain. 64 In contrast, vicriviroc seems to have better CNS penetration. 65 The reason for this discrepancy may be related to vicriviroc not being a substrate for p-glycoprotein and to its greater lipophilic properties compared to maraviroc.64,66,67 Clearly, additional studies are needed to examine and compare the pharmacokinetics of CCR5 antagonists in human CNS.
Clinical Experience with Maraviroc
HIV-1 Tropism Determination
Because CCR5 antagonists are active against R5 HIV-1 only, assessment of viral tropism is recommended prior to treatment with CCR5 antagonists. In patients failing treatment with maraviroc or vicriviroc, the main pathway of viral escape was the selection of preexisting CXCR4-using HIV-1 variants,68–70 further underscoring the importance of tropism determination. Tropism can be assessed by both phenotypic and genotypic assays. Traditionally, phenotyping was done in the MT-2 cell line, 71 which expresses CXCR4 but not CCR5.19,25,72 Because MT-2 culture assays can take several weeks, they may not be suitable for clinical use. Genotyping by population sequencing of the V3 region of Env from plasma virions, followed by tropism inference using an algoritm, such as geno2pheno (g2P) or PSSM, has faster turn-around times. 73 However, genotyping has a low sensitivity for detection of X4 variants. 74 As a result, clinical trials with CCR5 antagonists have generally used the Trofile assay (Monogram Biosciences, San Francisco, CA), a phenotypic assay that evaluates coreceptor use by Env sequences amplified from plasma by PCR. Following PCR amplification, complete or partial (V1 to V3) Env regions are cloned into an expression vector and used to generate pseudoviruses for infection of indicator cell lines expressing CD4 and CCR5 or CXCR4. The early version of the Trofile assay, which was used in the maraviroc trials, detected X4 variants with a 100% sensitivity but only when such variants represented≥10% of viral variants in the sample. Despite the superior sensitivity of Trofile compared to genotyping, preliminary data suggest that both the Trofile assay and genotyping are similarly effective at detecting CXCR4-using variants and thereby at predicting viral load reductions, at least in patients with multidrug-resistant virus. 75 Yet, the original Trofile assay is not sufficiently sensitive for detection of X4 viruses because patients initially classified as R5 HIV-1 actually contained low-frequency X4 variants, which emerged under treatment with CCR5 antagonists.68–70 There is currently an improved Trofile assay, called “enhanced Trofile”, with a sensitivity of 100% for detecting X4 viruses accounting for ≥0.3% of the virus population (Monogram Biosciences, San Francisco, CA). There are also improved genotyping methodologies using pyrosequencing, with reported detection of X4 variants representing 0.5% of the viral population.68,76 It will be critical to compare enhanced Trofile and pyrosequencing in detection of minor X4 variants in patients considering treatment with CCR5 antagonists.
Maraviroc Efficacy
Maraviroc efficacy, first evaluated clinically in a 10-day monotherapy trial in patients with R5 HIV-1, reduced viral loads by ≥1.6 log10. 58 These encouraging data led to the phase III clinical trials, MOTIVATE-1 and −2. MOTIVATE-1 was conducted in the United States and Canada, and MOTIVATE-2 in the United States, Australia and Europe. Both trials, with identical designs, evaluated the efficacy and safety of adding maraviroc (either once or twice daily) to Optimized Background Therapy (OBT) in a total of 1049 treatment-experienced patients without detectable X4 HIV-1 (determined by the original Trofile assay). Virologic response, defined as <50 copies HIV-1 RNA/ml plasma at 48 weeks, occurred in 47% of patients in the OBT plus twice-daily maraviroc group and 42% in the OBT plus once-daily maraviroc group versus 16% of those receiving OBT only (both P < 0.001). In addition, CD4+ count recovery was greater with maraviroc once or twice daily than with placebo (both P < 0.001).77,78
In a Phase II study similar to MOTIVATE but in patients with X4- or R5X4- HIV, there was no statistically significant differences in week 48 viral loads between the OBT plus maraviroc regimens versus OBT alone (−0.62 and −1.11 vs. −0.84 log10 HIV-1 RNA copies/ml, respectively) or change in CD4+ count (+65 and +78 vs. +51 cells/μl, respectively). 79 Thus, maraviroc offers no significant clinical benefit in patients with X4 or R5X4 HIV-1 viruses. 80
The efficacy of maraviroc in treatment-naïve patients was evaluated in the MERIT study, a Phase III trial comparing maraviroc and efavirenz, each in combination with zidovudine and lamivudine. 81 Patients with R5-only HIV-1 were randomized to zidovudine/lamivudine with either efavirenz or once- or twice-daily maraviroc. The once-daily maraviroc arm was closed because of inferior efficacy. The twice-daily maraviroc arm demonstrated similar results to the control, with 69.3% vs. 65.3% of patients having <50 HIV-1 RNA copies/ml. Yet, this small difference in response did not meet the predefined 10% criteria for noninferiority. It is likely that failure to demonstrate noninferiority was due to the relatively low sensitivity of the original Trofile assay used for screening of patients. A recent analysis of the data following tropism screening with the more sensitive “Enhanced Trofile assay” (see above), demonstrated that for those patients with a true R5-only phenotype, there were almost identical virological responses in the maraviroc and efavirenz treatments. 82
Maraviroc Safety
Maraviroc has consistently demonstrated a safety profile similar to that of placebo.58,77,78,80 The most common adverse effects are cough, pyrexia, infections of the upper respiratory tract, rash, musculoskeletal symptoms, lightheadedness, and abdominal pain. Importantly, maraviroc is not associated with cardiovascular events, hepatotoxicity or development of malignancies, all of which raised serious concerns in early clinical studies with other CCR5 antagonists. In this regard, the clinical development of aplaviroc ended because of hepatotoxicity 83 and that of Sch-C (a vicriviroc precursor) because of electrocardiographic QTc interval prolongation. 59 A Phase II trial suggested an association between vicriviroc and increased risk of malignancy, 7 but was not confirmed in a later study. 84 Although 11 patients (1.3%) in the maraviroc Phase III studies reported cardiovascular events such as myocardial ischemia, these patients had either heart disease or heart disease factors, precluding a clear association between maraviroc and cardiovascular toxicity. Likewise, out of 1300 patients enrolled in the Phase IIb/III studies, one had severe hepatotoxicity, but again, could not be associated with maraviroc because the patient was taking other medications with potential hepatotoxicity. Finally, the clinical data on maraviroc does not support an association with malignancy development. Overall, maraviroc safety profile is encouraging. However, given the potential for malignancies and cardiovascular and liver toxicities with CCR5 antagonist use, the Food and Drug Administration (FDA) requires long-term follow-up toxicity studies.
HIV-1 Resistance to Maraviroc and other CCR5 Antagonists
Resistance to CCR5 antagonists may arise from viral use of the alternative coreceptor CXCR4, either by acquiring mutations that allow switch to CXCR4 use or by selection of preexisting CXCR4-using variants. In vitro data indicate that CXCR4 switch under CCR5 antagonist pressure is rare. 85 In vivo, some patients failing treatment with maraviroc or vicriviroc were found to harbor X4 variants, but sequencing analysis demonstrated that such variants were most likely selected from minor populations already present prior to treatment.69,70 In MOTIVATE-1 and −2, approximately 5% of patients with R5 HIV-1 only at screening (4-6 weeks prior to the beginning of the trial) had evidence of X4 HIV-1 at trial entry. For this subset of patients, 27% of those receiving OBT plus maraviroc once daily, 18% of those receiving OBT plus maraviroc twice daily and 18% of those receiving OBT alone, had <50 HIV-1 RNA copies/ml at week 24. In contrast, among patients with R5 HIV-1 only at both screening and study entry, 50% in both OBT plus maraviroc groups and 26% in the OBT-alone group had <50 HIV-1 RNA copies/ml at week 24. That ~5% of patients classified as having R5 HIV-1 only at screening turned out to have X4 HIV-1 variants at study entry may reflect the limited sensitivity of the HIV-1 coreceptor tropism assay used (sensitivity of 100% for detection of X4 variants when such variants represented ≥10% of the viral population, see above). These data suggest that selection of preexisting, yet undetected, X4 HIV-1 variants may account for virologic failure in patients taking maraviroc.
Resistance to CCR5 antagonists can also arise from emergence of R5 HIV-1 variants with increased affinity for CCR5 (partial resistance) or from variants capable of infection via antagonist-bound CCR5 (full resistance) (Fig. 3). Partial and full resistance have been observed both in vitro85,86 and in vivo. 69 In genotypic assays, resistance is generally associated with mutations in Env, generally in the V3 region,69,87–89 but no signature mutations for resistance to CCR5 antagonist have been identified to date. Some of these mutant Envs are less dependent on interactions with CCR5 extracellular loops (mainly ECL2) but more dependent on interactions with the CCR5 N-terminus than wild-type Envs.90–92 In addition, there is in vitro evidence that full resistance to vicriviroc can be conferred by mutations in the fusion peptide of gp41 without changes in V3.88,93 Thus, resistance to CCR5 antagonists can follow both V3 dependent and V3 independent pathways. It will be important to determine the relative contribution of each resistance pathway in patients. Resistance to CCR5 antagonists is commonly diagnosed using the Phenosense Entry Susceptibility Assay (Monogram Biosciences), a single-cycle, Env-pseudotype assay based on U87 cells expressing high levels of CD4 and CCR5/CXCR4. In this assay, partial resistance is manifested by drug inhibition curves with increased values of EC50 (effective concentration that inhibits virus by 50%), whereas full resistance is manifested by incomplete dose response curves with inhibition plateaus at <100% inhibition.85,86 The height of the inhibition plateau in infection with fully resistant HIV-1 is indicative of the relative efficiencies with which free and antagonist-bound CCR5 are used, with greater inhibition plateaus indicating higher efficiencies in use of free CCR5. Currently, the factors determining the magnitude of inhibition plateaus in resistance phenotypic assays, and therefore the efficiency with which resistant viruses use antagonist-bound CCR5, are not well known. Elucidation of these factors is important because it will help understand resistance to CCR5 antagonists and its manifestation in phenotypic assays currently used in clinical studies. 94 We have recently demonstrated that reduced CCR5 density in lymphocytes (either in donors with low CCR5 levels or in donors treated with rapamycin) sensitizes R5 HIV-1 resistant to vicriviroc. 47 This impact of CCR5 density on antagonist activity against resistant HIV-1 was confirmed on cell lines with varying levels of CCR5 expression. These results represented the first indication that i) a host factor (CCR5 density) influences the way resistance to a CCR5 antagonist is manifested in a phenotypic assay, and ii) R5 HIV-1 strains that are fully resistant to a CCR5 antagonist recover drug sensitivity when CCR5 density is decreased, suggesting CCR5 reduction as an approach to control resistance.
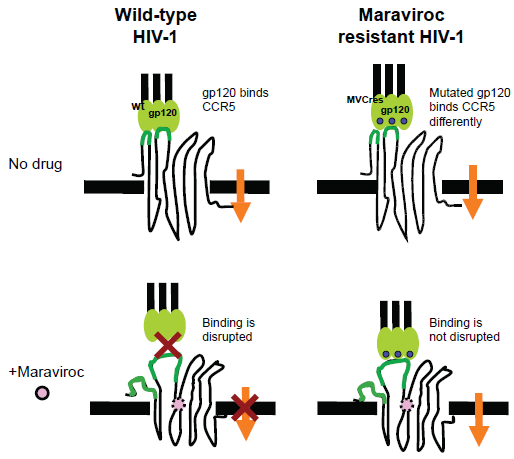
It is currently unclear whether resistance to maraviroc confers broad drug-class resistance.85,95 In one study, vicriviroc resistant viruses were resistant to other CCR5 antagonists (aplaviroc, maraviroc, AD101 and CMPD-167). 95 However, in another study, maraviroc-resistant HIV-1 was inhibited by aplaviroc. 85 It is possible that CCR5 antagonists may lock CCR5 in an antagonist-dependent conformation that is recognized by some, but not all, resistant viruses. Alternatively, aplaviroc inhibition of maraviroc-resistant HIV-1 might be explained by the rather unique aplaviroc binding to CCR5. Whereas most small-molecule antagonists have fewer interactions with CCR5 extracellular domains and insert deeply into the transmembrane region,32,33,96,97 aplaviroc binds in an almost horizontal position underneath the extracellular β-hairpin loop. As Phase III trials of vicriviroc advance, it will be critical to determine whether maraviroc-resistant clinical isolates can be inhibited by vicriviroc. In addition, the recently completed mapping of the CCR5 binding pocket could provide critical insights for structure-based design of novel CCR5 antagonists with activity against antagonist-resistant viruses.31,98
Potential Use of Maraviroc in Treatment-Naïve Patients and in Selected Settings
Potential Use of Maraviroc in Treatment-Naïve Patients
Maraviroc has favorable antiviral interactions (additivity or slight synergy) with NRTIs, NNRTIs, PIs and enfuvirtide, 15 and thus, incorporating maraviroc in drug regimens could increase viral suppression in patients. Although maraviroc is currently indicated for treatment-experienced patients carrying R5 HIV-1 strains, anticipated results from a Phase II vicriviroc study (initiated in January, 2008) could recommend vicriviroc, and perhaps maraviroc, for treatment-naïve patients. 99 The trial initially included 95 treatmentnaïve patients with R5 HIV taking vicriviroc plus ritonavir-boosted atazanavir vs. Truvada (emtricitabine and tenofovir, two NRTIs) plus ritonavir-boosted atazanavir. Following a formal interim analysis and safety data review at week 24, the study was expanded to 105 additional patients. The primary efficacy endpoint will be the mean change from baseline in viral loads at week 48. Should vicriviroc demonstrate non-inferiority compared to Truvada, the data will suggest that vicriviroc, and possibly maraviroc, could offer a new first-line therapy option in treatment-naïve patients. Because 80% to 90% of treatment-naïve patients carry R5 HIV-1 strains only, 100 first-line therapy with CCR5 antagonists could benefit many patients by preserving other antiretroviral classes for later treatment.
Potential Use of Maraviroc in Prevention of HIV-1 Transmission
To date, topical administration of the CCR5 antagonist CMPD-167 or RANTES analogs (PSC-RANTES, 5P12-RANTES and 6P4-RANTES) has demonstrated efficacy in a macaque model of vaginal R5-tropic SHIV-1 transmission.101–103 Vaginal transmission was prevented (8/10 and 5/5 protected animals for CMPD-167 and for each RANTES analog, respectively) but only at high doses (5 mM CMPD-167 and 1 mM RANTES analog), probably due to drug delivery pharmacology. 104 Encouraging data demonstrate that antiviral synergy by different inhibitors prevents viral transmission at lower effective doses. 105
Oral administration of CMPD-167 has shown efficacy in preventing R5-SHIV-1 transmission, but only at doses higher than those used topically. 106 Importantly, however, oral administration of maraviroc gives similar drug concentrations in plasma and cervicovaginal fluid, suggesting that achievable maraviroc doses might prevent viral transmission. 107 Thus, the potential use of maraviroc, perhaps in synergistic combinations with other entry inhibitors, in pre- and post-exposure prophylaxis against HIV-1 transmission requires further investigation.
Potential Use of Maraviroc in Solid-Organ Transplantation in HIV-1 Infection
Since the introduction of HAART, HIV-1 patients are living longer and comorbidities such as heart, liver and kidney disease are becoming serious medical problems.108,109 Among HIV-1 patients living in New York City, 22% of African American and 11.4% of Whites have either chronic kidney disease or endstage renal disease. 110 In France, the proportion of HIV-1 patients dying of end-stage liver disease has increased from 2% in 1995 to 17% in 2005. 111 As a result of increased morbidity and mortality rates in HIV-1 patients with end-stage organ disease,112,113 many transplant centers have begun to transplant organs into selected patients.114–116 Transplant patients receive both antiretrovirals and immunosuppressants (commonly cyclosporine, tacrolimus, mycophenolic acid and rapamycin), but there is no consensus on drug combinations.115,117,118 The combinations of mycophenolic acid and zidovudine (AZT) or stavudine (D4T) are generally avoided 118 due to antiviral drug antagonism. 119 Because many immunosuppressants are metabolized by the cytochrome P450 (CYP450) enzyme system (mainly through the CYP3A4 isoform), 120 which is inhibited by PIs and induced by NNRTIs, 121 co-administration of immunosuppressants and PIs/NNRTIs often results in significant drug interactions. These drug interactions can perturb drug levels and thereby lead to toxicity, insufficient immunosuppression and reduced HIV-1 control. 118 For these reasons, close monitoring and adjustment of drug levels in patients treated with immunosuppressants and HAART are of critical importance. 118 Since maraviroc and other CCR5 antagonists are neither inhibitors nor inducers of CYP3A4, their use in transplant patients may prevent potentially harmful drug interactions resulting from altered immunosuppressant blood levels. In addition, combinations of immunosuppressants and CCR5 antagonists will have lesser effects on blood levels of CYP3A4-substrate non-HIV-1 medications (many macrolide antibiotics, statins and psychotropic drugs) for the treatment of comorbidities especially frequent in older patients. In this regard, a retrospective study of HIV-1 patients older than 55 in New York City found that 89% had comorbidities and 81% were taking non-HIV medications. 122
A recent analysis of 100 HIV-1 patients receiving kidney transplants demonstrated 1-year patient survival was similar between HIV-1-infected and uninfected groups (95.4% and 96.2%, for infected and uninfected, respectively; P = 0.32). 123 However, 1-year organ survival was significantly lower for HIV-1 patients (87.9% vs. 94.6%; P = 0.03). After subsequent subgroup analyses, the different outcomes were explained by several risk factors, especially older donor age and delayed graft function (DGF) in HIV-infected recipients. As in previous studies,114,124 the increased susceptibility of HIV-1 transplant patients to the detrimental effects of DGF was related to nephrotoxicity by combinations of calcineurin inhibitors and PIs. Because CCR5 antagonists, unlike PIs and NNRTIs, do not alter calcineurin inhibitor concentrations, 125 their use could provide a safer antiretroviral option. In addition, lower transplant rejection rates in individuals lacking CCR5 expression (CCR5δ32 homozygous) 126 or following CCR5 blockade,127,128 further supports the idea that CCR5 antagonists will prolong survival of the transplanted organ in HIV-1 patients.
Potential Use of Maraviroc in other Settings
As recently reviewed by Soriano et al, 129 maraviroc may provide a convenient antiretroviral option for HIV-1 patients with an increased risk for heart disease. Commonly used PIs and NRTIs (most notably abacavir and didanosine) are associated with increased risk of cardiovascular events,130,131 and thus, switching to a maraviroc-containing regimen could be beneficial for those patients with both R5 HIV-1 and increased risk for heart disease. Likewise, since HIV-1 entry inhibitors are associated with a recovery of CD4+ counts even in the absence of complete viral suppression, maraviroc could serve as a strategy for immunological restoration in selected patients.132,133 It is also possible that maraviroc will improve treatment options for HIV-1 patients with tuberculosis and for those coinfected with hepatitis viruses. 129
Summary
The recently developed antiretroviral classes of entry and integrase inhibitors offer much-needed treatment options for patients with drug-resistant HIV-1 infection. The fusion inhibitor enfuvirtide and the small-molecule CCR5 antagonist maraviroc are the first two licensed entry inhibitors, while vicriviroc and the CCR5 antibodies PRO 140 and HGS004 are in clinical trials.7,8,134 Entry inhibitors are currently recommended for patients with drug-resistant HIV-1. Favorable pharmacokinetic and antiviral interactions between entry inhibitors and drugs from the NRTI, NNRTI, PI and integrase inhibitor classes suggest that their combinations will be conveniently administered.15,59 Importantly, combinations of small-molecule CCR5 antagonists and CCR5 antibodies exhibit potent antiviral synergies, probably explained by their binding to different regions of CCR5 and thereby disruption of sequential steps on viral entry.135–138 Due to coreceptor specificity, the use of CCR5 inhibitors is limited to patients lacking X4 viruses. However, since 50% to 62% of patients with drug-resistant HIV-1 carry R5 HIV-1 only, 139 maraviroc and other CCR5 inhibitors will benefit a majority of the treatment-experienced HIV-1 infected population. Moreover, with expanded access of antiretroviral therapy to other countries, CCR5 inhibitors could be particularly effective against Subtype C HIV-1, which rarely switches to CXCR4 coreceptor use, accounts for >50% infections worldwide, and is the most prevalent subtype in sub-Saharan Africa and parts of Asia.56,140
In addition, maraviroc (and perhaps other CCR5 inhibitors) could improve treatment for the increasing numbers of older HIV-1 patients with organ failure receiving organ transplants.115,141 Because maraviroc does not inhibit or induce CYP3A4, combinations of maraviroc and transplantation immunosuppressants have low potential for organ rejection secondary to insufficient immunosuppression or for toxicity secondary to overdosing. Of note, our results demonstrate that selected immunosuppressants reduce CCR5 expression and have synergistic antiviral activities with both enfuvirtide and CCR5 antagonists.44,46,47,142 Moreover, CCR5 antagonists prevent acute and chronic rejection of transplanted organs,127,128 providing an additional potential benefit for HIV-1 infected transplant recipients. Prevention of vaginal SHIV transmission in animal models by CCR5 inhibitors,101–103,105,106 coupled with achievement of high levels of maraviroc in cervicovaginal fluid following oral administration, 107 suggest that maraviroc could curtail sexual transmission of HIV-1, which is virtually always driven by R5 strains. 143
Finally, results from a vicriviroc trial in treatmentnaïve HIV-1 patients, could lead to the use of CCR5 antagonists as a new first-line therapy, preserving drugs from other classes for later treatment. 99
Disclosures
The authors report no conflicts of interest.
Footnotes
Acknowledgements
We thank Dr. Robert R. Redfield and Dr. Robert C. Gallo for their continual mentoring and support. This project was supported in part by NIH grant AI084417 to A.H.