
Abstract
The evaluation and management of pulmonary arterial hypertension (PAH) is a rapidly evolving area of subspecialty medicine requiring regular clinical updates. Most notably are changes in the World Health Organization diagnostic scheme whereby the clinician categorizes the correct type of pulmonary hypertension in order direct the most specific evaluation and treatment plan. In addition, there have been several changes in both the FDA-approved pharmaceutical formulations and new agents for the treatment of PAH. This review will provide an update in these areas and more importantly, guidance to the clinician on the most appropriate utilization of these new treatment options.
Introduction
Pulmonary arterial hypertension (PAH) occurs due to restricted flow through the pulmonary arterial circulation resulting in increased pulmonary vascular resistance. As the condition worsens, right heart failure ensues. The pathophysiology is unclear but may involve multiple pathogenic pathways. 1 There is an imbalance of vasomediators and hemostasis favoring vasoconstriction and thrombosis in-situ. In addition, genetic and molecular derangements predispose and perhaps cause abnormal proliferation and function of both endothelial and vascular smooth muscle cells. Recent changes in the World Health Organization (WHO) diagnostic classification scheme and the FDA approval of new treatments for PAH have established the need for a clinical update. 2
Diagnosis
The patient will often present with cardiopulmonary symptoms, typically dyspnea, prompting medical evaluation. The combination of syncope and dyspnea should raise the clinical suspicion of PAH. In addition, appropriate risk factor setting such as a history of prolonged use of dietary suppressants or the presence of collagen vascular disease, particularly scleroderma, should also prompt strong consideration of the diagnosis. The wide-spread availability of echocardiography and non-invasive nature of the test has secured a role in the evaluation of dyspnea. Right ventricular systolic pressures are generally estimated using the peak tricuspid regurgitant jet velocity in meters per second and equate to the pulmonary artery systolic pressure. Newer methods have been developed to measure and/or estimate the mean pulmonary artery pressure (PAP). 3 The mean PAP defines the disease and is a stronger predictor of outcome than pulmonary artery systolic pressure. 4 Nonetheless, confirmation of the diagnosis requires right heart catheterization (RHC). Specific measurements of the right heart and PAP are used with the cardiac output (CO) to determine the pulmonary vascular resistance (PVR). Mean PAP in excess of 25 mmHg and PVR ≥ 3 Woods units define PAH. The RHC affords three other components important in the assessment of PAH: exclusion of other diagnoses, determination of prognosis and assessment of acute vasoreactivity. Conditions to be excluded include left-to-right shunting by virtue of an oxygen saturation run as the catheter progresses from the systemic venous circulation into the right heart and pulmonary artery. Elevated left heart pressures (pulmonary artery occlusion “wedge” or left-ventricular end-diastolic pressure >15 mmHg) indicate pulmonary venous hypertension, not PAH. Severe elevations in right heart pressures, particularly right atrial pressure, and low cardiac index indicate a poorer prognosis. 4 Lastly, acute vasoresponsiveness can be assessed by the administration of a short-acting pulmonary arterial vasodilator (intravenous epoprostenol or adenosine versus inhaled nitric oxide) and remeasurement of the mean PAP and CO. The mean PAP should drop by at least 10 to below 40 mmHg with preserved or increased CO. 5 Once the RHC confirms the presence of PAH, then other testing may be necessary to exclude co-morbid conditions. For example, a ventilation-perfusion lung scan is recommended to exclude chronic thromboembolic disease as the cause of the pulmonary hypertension (PH). Computed tomography with contrast can be falsely negative. 6 This variant of PH is especially important to identify as the treatment of choice is a pulmonary endarterectomy in eligible patients. The 2008 WHO conference on PAH has resulted in an updated classification scheme for the various diagnoses associated with PH (Table 1). This diagnostic classification enables the clinician to be more precise in the designation of the various types of PH and to guide the evaluation for the multiple co-morbid conditions.
New World Health Organization Diagnostic Groups (Dana Point, 2008).

The World Health Organization Consensus Conference on pulmonary hypertension in Dana Point, California 2008 with the major changes from the 2003 Venice classification highlighted in bold.
Adapted from Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension.
Treatment
The American College of Chest Physicians evidenced-based guidelines for PH were first published in 2004. 7 The treatment guidelines were subsequently updated in 2007 8 with additional modification by the American Heart Association. 9 Since the 2007 update, 2 new pharmaceutical agents and 2 new delivery systems have been approved by the FDA. In addition, the indications for use of bosentan have been broadened to include WHO functional class (FC) II (Table 2). The evidence for the newer agents and rationale for the other changes will be discussed. The newer options require modification of the treatment guidelines (Tables 2 and 3).
Currently FDA-approved therapies for pulmonary arterial hypertension.

The currently available, FDA-approved therapies are listed according to the approved World Health Organization functional class indication for each drug. Note that most drugs are approved for use in multiple classes. The new drugs or indications are highlighted (see table 2 for additional detail).
Newly approved pulmonary arterial hypertension therapies.

Several changes in FDA-approved therapies in 2009 are described.
Currently, there are 4 oral, 2 inhaled and 2 infusion agents that are FDA-approved for treatment of PAH in the United States (Table 2). In addition, calcium-channel blockers have been used with survival benefit in those patients with acute vasoresponsiveness.
10
The other oral agents represent 2 separate classes of medications: endothelin-receptor antagonists (ERA) and phosphodiesterase-5 (PDE-5) inhibitors. The first oral agent to be approved was bosentan, a dual ERA, which significantly increased six-minute walk distance (6MWD) in the BREATHE-1 trial.
11
Since the evidence-based treatment guideline was updated, a second ERA, more selective for receptor A, has been approved based on the Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Study 1 and 2 (ARIES-1 and ARIES-2) trials that demonstrated similar improvements in 6MWD.
12
Those trials were co-existing Phase 3 studies (ARIES 1 and 2) based in North America and Europe respectively. In all, 202 and 192 patients with PAH were randomized to receive either placebo or ambrisentan (ARIES-1, 5 or 10 mg; ARIES-2, 2.5 or 5 mg) orally once daily for 12 weeks. The primary end point for each study was change in 6MWD from baseline to week 12. Clinical worsening, WHO FC, Short Form-36 Health Survey score, Borg dyspnea score, and B-type natriuretic peptide (BNP) plasma concentrations also were assessed. In addition, a long-term extension study was performed. The 6MWD increased in all ambrisentan groups; mean placebo-corrected treatment effects were 31 meters (
Bosentan was originally approved for WHO FC III and IV while ambrisentan was approved for class II and III. The indication for bosentan was expanded in 2009 to include earlier stages of disease based on WHO FC as a result of the EARLY trial. 14 In that study, 185 patients were randomly assigned to receive bosentan (n = 93) or placebo (n = 92) for a 6-month double-blind treatment period. Primary endpoints were PVR at six months, expressed as percentage of baseline, and 6MWD, compared to baseline. The final data analysis was performed using 168 patients (80 in the bosentan group, 88 in the placebo group) for PVR and with 177 (86 and 91) for 6MWD. At the end of the 6-month study period, the geometric mean PVR was 83% [95% Confidence Interval (CI) 74–94] of the baseline value in the bosentan group and 108% (98–118) of the baseline value in the placebo group (treatment effect -24%, 95% CI -34 to -10; P < 0.0001). The mean 6MWD increased from baseline in the bosentan group (11 m, 95% CI -5 to 27) but decreased in the placebo group (-8 m, -24 to 9), with a modest mean treatment effect of 19 m (95% CI 4–42; P = 0.0758). Twelve (13%) patients in the bosentan group and eight (9%) in the placebo group reported serious adverse events, the most common of which were syncope in the bosentan group and right ventricular failure in the placebo group. If treatment of earlier stage disease is beneficial as suggested by the EARLY trial, then screening high risk patients for PH may be more beneficial. 15
Sildenafil was the first PDE-5 inhibitor to be approved for the treatment of PAH based on the Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study.
16
Although there seemed to be some modest dose effect, there was no difference in meaningful clinical events in this short-term study between 20 mg, 40 mg or 80 mg three times daily. Consequently, only the smallest dose was approved by the FDA in 2005. Unfortunately, the FDA-approved dose may the only one covered by insurance for some patients preventing dose titration upward in patients with only partial clinical response. Tadalafil, a longer-acting PDE-5 inhibitor requiring only once daily dosing, was approved in 2009 based on the Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study.
17
In this 16-week, double-blind, placebo-controlled study, 405 patients with PAH (idiopathic or associated), either treatment-naive or on background therapy with bosentan, were randomized to placebo or tadalafil 2.5, 10, 20, or 40 mg orally once daily. The primary endpoint was the change from baseline to week 16 in the 6MWD. Changes in WHO FC, clinical worsening, and health-related quality of life were also assessed. Patients completing the 16-week study could enter a long-term extension study. Tadalafil increased the 6MWD in a dose-dependent manner; however, only the 40-mg dose met the prespecified level of statistical significance (
Monthly costs of selected pulmonary arterial hypertension therapies.
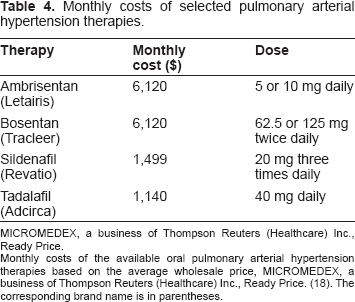
MICROMEDEX, a business of Thompson Reuters (Healthcare) Inc., Ready Price.
Monthly costs of the available oral pulmonary arterial hypertension therapies based on the average wholesale price, MICROMEDEX, a business of Thompson Reuters (Healthcare) Inc., Ready Price. (18). The corresponding brand name is in parentheses.
There have been recent changes in prostacyclin therapy that merit discussion, particularly with the inhaled formulations. Iloprost, an inhaled analogue of prostacyclin, was FDA-approved in 2004 based on a randomized, placebo-controlled, 12-week study, comparing iloprost to placebo. 19 In the final analysis, there was a placebo-corrected increase in 6MWD of 36 meters in 207 patients with symptomatic idiopathic PAH, PAH associated with connective tissue disease or appetite suppressants, or PH related to inoperable chronic thromboembolic disease. Due to the short half-life, the drug must be administered every 2 hours. Until recently the drug delivery system did not permit a very short treatment duration. In 2009, the FDA approved a more concentrated solution which may reduce the delivery time to less than 5 minutes. In addition, there is now a second option for inhaled prostacyclin therapy based on the Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension (TRIUMPH-1) was a randomized trial in which investigational inhaled treprostinil or placebo was added to either bosentan or sildenafil in 235 patients with PAH. 20 Patients taking bosentan and/or sildenafil plus treprostinil showed significantly more improvement in 6MWD (20 meters) than did those taking bosentan and/or sildenafil plus placebo. Inhaled treprostinil was approved in 2009 (Table 2). The administration is four times daily and the dose is gradually titrated upward to a maximum of 40 micrograms per dose. It appears well tolerated over the 24 month study period. The most common side effects were cough (54%), headache (38%) and nausea (21%).
Combination therapy remains controversial but may be considered in patients who do not adequately respond to monotherapy. 8 There is only modest literature is this area; however, combination therapy appears to be quite common. 21 In support of such therapy, the benefit of adding sildenafil to stable patients on intravenous epoprostenol was shown in the Pulmonary Arterial Hypertension Combination Study of Epoprostenol and Sildenafil (PACES-1) trial. 22 In that study, 267 PAH patients on a stable epoprostenol dose were randomized to receive either sildenafil or placebo. The patients taking epoprostenol and sildenafil showed a significant improvement in 6MWD after 16 weeks. Most patients in the combination therapy arm were taking sildenafil 80 mg three times daily. The potential limited availability of this higher dose is discussed above. An ongoing Phase IV trial entitled “Effects of combination of bosentan and sildenafil versus sildenafil monotherapy on morbidity and mortality in symptomatic patients with pulmonary arterial hypertension (COMPASS 2)” is nearing complete enrollment. This prospective, multicenter, double–-blind, randomized, placebo–-controlled trial will examine significant clinical morbidity such as hospitalization for PAH as will as mortality. This study will be important in the evaluation of the potential efficacy of combined oral therapies. Of note, a open-label study (ATHENA-1) examining the hemodynamic effect of the combination of ambrisentan and sildenafil compared to sildenafil only is also in progress.
Summary
Several important changes have occurred in the last 2 years that significantly alter the landscape of the evaluation and treatment of PAH. The diagnostic classification scheme was updated in 2008 at the WHO Consensus Conference and now includes several new subcategories that provide the clinician a more clinically relevant reference (Table 1). In addition, there have several recent developments in FDA-approved therapies for PAH. Most notably, tadalafil, a long-acting phosphodiesterase inhibitor, offers a once-daily and less expensive oral option. In addition, treprostinil is now approved for inhaled use at frequency that is less than iloprost. Bosentan has received an expanded indication to WHO FC II patients based on evidence that treatment in early stage disease improved PVR. A more concentrated solution of iloprost should decrease administration times improving patient satisfaction and perhaps compliance. Lastly, more data is now available in support of combination therapy but additional studies are in progress which will provide important information on the potential efficacy of combination oral therapy.
Disclosures
This manuscript has been read and approved by the author. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The author reports no conflicts of interest.