
Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease leading to obstruction of the small pulmonary arteries. Currently, there are eight approved medications for PAH. This review article focuses on the mechanisms, clinical trials, efficacy, and safety profiles of each of the PAH medications. In addition, this review addresses combination PAH therapy, patient preference, and future therapies.
Introduction
The field of pulmonary vascular disease has grown substantially in the past several decades, and currently there are eight therapies approved for pulmonary arterial hypertension in the United States. Pulmonary hypertension, defined as a mean pulmonary artery pressure greater than 25 mmHg, is a broad term describing any elevation in pulmonary artery pressure. In order to enhance patient communication and facilitate homogenous enrollment in trials and treatment recommendations, pulmonary hypertension is subdivided into five groups based on the 2008 Dana Point World Symposium on Pulmonary Hypertension (Table 1.) 1
Clinical classification of pulmonary hypertension. 1
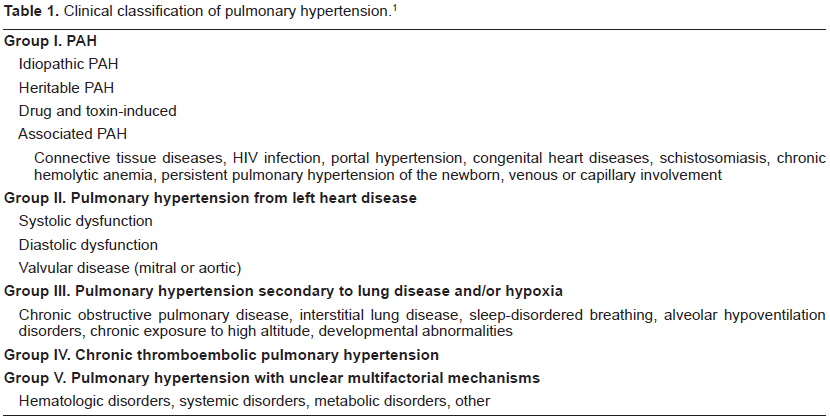
Group I includes idiopathic pulmonary arterial hypertension (IPAH), heritable pulmonary arterial hypertension (HPAH), drug and toxin induced PAH, and pulmonary hypertension related to risk factors or associated conditions (APAH) such as collagen vascular disease, HIV infection, portal hypertension, congenital systemic-to-pulmonary shunts, schistosomiasis, chronic hemolytic anemia, persistent pulmonary hypertension of the newborn, and venous or capillary involvement. Group II includes pulmonary hypertension associated with left-sided heart disease i.e. systolic dysfunction, diastolic dysfunction, aortic and mitral valvular disease. Group III includes pulmonary hypertension secondary to lung disease and/or hypoxia, Group IV pulmonary hypertension is secondary to chronic thrombotic and/or embolic disease, and Group V is a miscellaneous group. 1 This article will focus on Group I pulmonary hypertension, referred to collectively as pulmonary arterial hypertension (PAH).
PAH is a progressive disease characterized by narrowing of the pre-capillary pulmonary arteries. Obstruction of these distal pulmonary arteries leads to increased pulmonary vascular resistance and eventually right heart failure and death. Histological changes of PAH include endothelial cell proliferation, smooth muscle hypertrophy, adventitial changes, and obstruction of small pulmonary arteries. Late stages may show plexiform lesions and recanalization of vessels.
In 2000, mutations in bone morphogenic protein receptor type 2
Patients with PAH can present with fatigue, shortness of breath, angina, and syncope. They may have signs of right-sided heart failure including elevated jugular venous pressure, right-sided heave, systolic murmur over the left sternal border, pronounced second heart sound, ascites, and edema. Unfortunately, by the time PAH patients develop symptoms, they have irreversibly lost greater than 60% of their peripheral pulmonary vasculature. Although many patients with pulmonary hypertension are first identified by Doppler echocardiogram, the diagnosis of PAH requires right heart catheterization. Right heart catheterization is invasive, but a relatively safe procedure; in a combined retrospective and prospective evaluation of serious adverse events in pulmonary hypertension patients related to right heart catheterization, the number of serious adverse events was 76 out of 7,218 (1.1%), and there were only four fatal events resulting in an overall procedure-related mortality of 0.055%. 5 In order to diagnose a patient with PAH, left atrial pressure should be documented to be ≤15 mmHg, thus measurement of pulmonary capillary wedge pressure is an important surrogate for left atrial pressure to exclude pulmonary venous hypertension. Patients with elevated pulmonary capillary wedge pressure generally do not benefit from PAH-specific medications discussed in this review. In addition, right heart catheterization provides important prognostic information such as cardiac index (CI) and right atrial pressure and allows specialized procedures such as a fluid challenge.6,7 In patients with PAH, vasodilator testing, reviewed in a later section, is critical to determining therapeutic options.
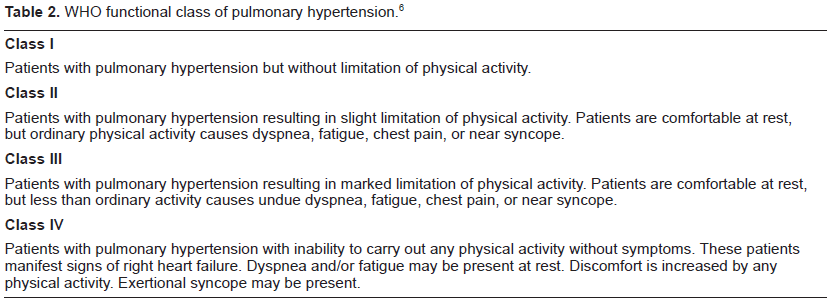
Six-minute walk distance (6-MWD) is a readily available assessment of cardiopulmonary exercise capacity and has been shown to correlate well with mortality. 8 Despite the many limitations, this test has served as the primary endpoint in most major therapeutic trials in PAH. Functional status is classified based on the World Health Organization (WHO) system (Table 2). A comprehensive clinical picture incorporating patient history, physical examination, echocardiography data, exercise capacity, and invasive hemodynamic measurements allows determination of therapeutic recommendations for PAH.
WHO functional class of pulmonary hypertension. 6
Over the past two decades there have been major advances in the treatment of PAH, and updated treatment guidelines have recently been published. 9 Traditional therapies for PAH including oxygen for hypoxic patients, diuretics in patients with right-sided heart failure, anticoagulation in the absence of contraindications, and inotrops in decompensated right heart failure are still prescribed in context. However, physicians now have additional therapies including calcium-channel antagonists, prostanoids, endothelin antagonists, and phosphodiesterase type 5 inhibitors (PDE5-I). Prior to the above therapies, patients had an average life span of 2.8 years after diagnosis. 6 Current therapies have improved symptoms, hemodynamics, and even survival; however, there is still no cure for PAH. 10
This review article focuses on the mechanisms, clinical trials, efficacy, and safety profiles of each of the PAH mediations. In addition, we will comment on combination PAH therapy, patient preference, and future therapies.
Prostacyclins
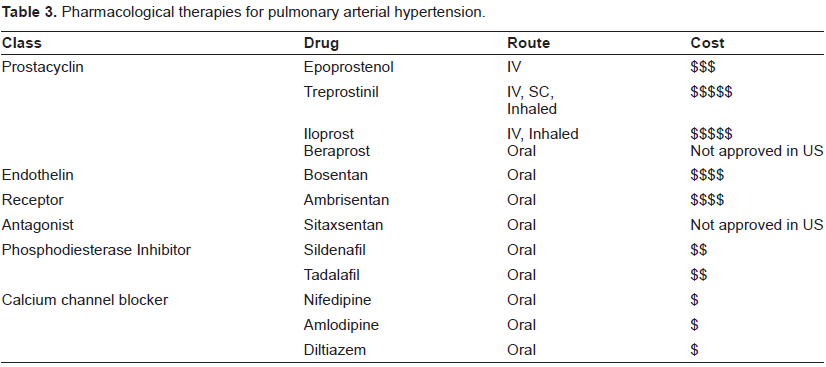
Prostacyclin, a metabolite of arachidonic acid, is produced in vascular endothelium, is potent systemic and pulmonary vasodilator, and has antiplatelet aggregation properties. Prostacyclin leads to vascular smooth muscle relaxation via binding to a G-protein-coupled, activating adenylate cyclase, and increasing production of intracellular cyclic AMP. Prostacyclin is a potent inhibitor of platelet aggregation and thrombus formation as well as inhibition of vascular cell proliferation. 11 Relative prostacyclin deficiency is thought to contribute to PAH. Patients with pulmonary hypertension were found to have decreased prostacyclin metabolite excretion compared with controls. 12 Treatment with prostacyclin analogues has made a dramatic impact on PAH and offers the best options for patients with advanced PAH. Prostacyclin therapy can be admini stered by intravenous (IV) (epoprostenol, treprostinil, iloprost), subcutaneous (treprostinil), inhalation (iloprost, treprostinil) or oral (beraprost, treprostinil) routes (Table 3).
Pharmacological therapies for pulmonary arterial hypertension.
Continuous intravenous epoprostenol was first used to treated PAH in 1984 and approved by the FDA in 1995 for functional class III-IV. 13 Epoprostenol is a potent vasodilator of both the systemic and pulmonary arteries. In addition, epoprostenol has antithrombotic properties related to effects on platelets, inhibits smooth muscle proliferation, and has positive inotropic properties.11,14 In 1996, a 12 week randomized prospective trial demonstrated that continuous intravenous infusion of epoprostenol increased 6-MWD, decreased mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR), and increased survival compared with conventional therapy alone.8,13 One hundred sixty-two PAH patients with NYHA class III-IV heart failure treated with epoprostenol were followed for a mean of 36 months. Survival with epoprostenol therapy was 88%, 76%, and 63% at one, two, and three years respectively; significantly better than the predicted survival rates at 59%, 46%, and 35% based on historical data.6,10 This improvement in survival in the initial epoprostenol trial and subsequent follow-up data has made epoprostenol the “gold standard” in treatment of PAH against which all other therapies are measured. Despite the excellent benefit offered by this drug, epoprostenol therapy is complex; its short half-life of less than 6 minutes requires continuous intravenous infusion through a permanent tunneled catheter. The medication must be refrigerated during administration; therefore patients must wear a cooled portable infusion pump. Rebound pulmonary hypertension may occur with interruptions in infusion. Minor complications of epoprostenol include jaw pain, diarrhea, flushing, headaches, nausea, and vomiting. Serious complications include catheter-related sepsis, thrombosis, and malfunction of the drug-delivery system. 8 At our institution, epoprostenol is initiated during an inpatient hospitalization at a dose of 1 ng/kg/min and increased by 1 ng/kg/min every 12 hours until side effects limit further titration or a dose of about 4-6 ng/kg/min is achieved. The drug is then increased once or twice a week by 1 ng/kg/min. Patients are re-evaluated at one to two months after starting IV epoprostenol therapy. The dose is further titrated to improve symptoms without prohibitive side effects and to obtain a normal cardiac index. The dose is usually maintained at 20-30 ng/kg/min. Unfortunately, there is a subset of patients who do not respond to epoprostenol therapy.
Treprostinil, a tricyclic benzidine analog of epoprostenol, was approved for PAH by the FDA in 2004, and is available for subcutaneous, IV, and inhaled use and is currently in oral trials. Unlike epoprostenol, it is chemically stable at room temperature and has a longer half-life of three to four hours. In a 12-week double-blind, placebo-controlled multicenter trial in 470 patients, subcutaneously administered treprostinil improved 6-MWD, Borg dyspnea score, mPAP, PVR, CI, and quality of life compared with controls.
15
Side effects include headache, diarrhea, nausea, rash, jaw pain, and over 80% of patients had infusion site pain/reaction.
15
The safety and efficacy of IV treprostinil was investigated in PAH in a 12-week open-label uncontrolled trial in 16 patients with PAH. Patients reached a mean dose of 41 +/- 4 ng/kg/min. Compared with controls, patients receiving treprostinil had an improvement in their 6-MWD, Borg dyspnea score, WHO functional class, mPAP, PVR, CI, and had no serious adverse events.
16
Interestingly, IV treprostinil appears to have a an increase in gram-negative bloodstream infections including
Iloprost is a prostacyclin analogue administered via inhalation. The drug is delivered by an ultrasonic or jet nebulizer device. Secondary to its short halflife, around one hour, iloprost requires frequent administration six to nine times a day. In a 12-week multicenter study administering iloprost six to nine times daily, there was improvement in the 6-MWD, NYHA functional class, and time to clinical deterioration without an increase in serious adverse events. 21 Inhaled iloprost was approved by the FDA in 2004 for class III-IV PAH patients. Lastly, beraprost, an oral prostacyclin analogue, is available in Korea and Japan after demonstrating improvement in 6-MWD at six months; however the effect was not evident at 9 or 12 months. 22 A substantial proportion of patients administered this drug have intolerable limb pain. The yearly cost for epoprostenol is $33,153, treprostinil $97,615, and iloprost $92,146. 9
Endothelin Receptor Antagonists
Endothelin-1 (ET-1) is a potent vasoconstrictor and smooth muscle mitogen that is over expressed in the plasma and lung tissue of patients with PAH. 23 There are two ET-1 receptor isoforms, ETA and ETB. Activation of ETA causes vasoconstriction and proliferation of vascular smooth muscle cells while ETB activation leads to clearance of ET-1 and a compensatory vasodilator response. 24 It currently unknown whether it is preferable to block both ETA and ETB receptors, or to selectively target the ETA receptor. Two of the three oral endothelin receptor antagonist are FDA approved, bosentan, which blocks both ETA and ETB, and ambrisentan, a selective ETA blocker. Sitaxsentan is also a selective ETA receptor antagonist and is approved in Europe, Canada, and Australia. The STRIDE-1 and STRIDE-2 studies demonstrated improvement in 6-MWD with 100 mg/day of sitaxsentan, and one year safety and efficacy was demonstrated in the STRIDE-2X study. 25
Bosentan was the first oral therapy approved by the FDA for the treatment of PAH. In 2001, a small double-blind placebo-controlled study in 32 patients receiving bosentan 125 mg twice daily versus placebo demonstrated an improvement in 6-MWD, mPAP, CI, and WHO functional class in patients receiving bosentan. 26 A second double-blind, placebo-controlled study of 213 patients with PAH who received placebo or 62.5 mg of bosentan twice daily for 4 weeks followed by either of two doses of bosentan 125 or 250 mg twice daily for a minimum of 12 weeks demonstrated an improvement in 6-MWD, Borg dyspnea index, WHO functional class, and increased time to clinical worsening. 27 Abnormal hepatic function levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) occurred more frequently in the bosentan treated patients than in placebo and was found to be dose-dependent. Elevations over three times the upper limit of normal occurred in 4% of patients receiving 125 mg and 14% receiving 250 mg. Three patients had persistent elevations of liver enzymes and were withdrawn prematurely from the study. 27 Liver function improves with dose decrease or discontinuation. The most likely mechanism for the liver enzyme abnormalities is a dose-dependent competition of bosentan with the biliary excretion of bile salts resulting in retention of bile salts that can be cytotoxic to hepatocytes. 28 Secondary to common hepatic side effects, liver function test should be checked monthly.
Ambrisentan, a selective ETA antagonist, has high oral bioavailability 90% and maximum plasma concentration levels between 1.7 and 3.3 hours after administration. 29 The pharmacokinetics are doselinear. Steady state is achieved after 3-4 days and the half-life is approximately 15 hours. 29 Ambrisentan is excreted mostly unchanged in bile. A phase II trial in 64 patients with PAH demonstrated an increase in 6-MWD after 12 weeks of therapy for 1, 2.5, 5, or 10 mg once-daily dosing. There were also improvements in Borg dyspnea index score and WHO function class. A subgroup of 29 patients had hemodynamic testing showing a decrease in mPAP and PVR and increase in CI compared with baseline. Two out of 64 patients had elevation in LFTs > 3 times the upper limit of normal during the first 24 weeks of therapy. 29 A combined analysis demonstrated that the clinical benefits of ambrisentan were sustained over the 1-year study period. 29 Two concurrent, randomized, double blind, placebo-controlled phase III trials followed. In ARIES-1 (US and Australia), patients with PAH were randomized to 5 mg or 10 mg ambrisentan or placebo daily while in ARIES-2 (Europe, Africa, and South America) patients were randomized to 2.5 or 5 mg ambrisentan or placebo daily. Ambrisentan improved 6-MWD at all doses, and at the 5 mg/day dose delayed time to clinical worsening. None of the 261 patients in ARIES-1 and ARIES-2 developed serum ALT or AST > 3 times upper limits of normal. 30 Therefore, patients receiving ambrisentan have a lower incidence of elevated liver function tests than bosentan or sitaxsentan in clinical trials. Side effects of both endothelin receptor antagonists include nasal congestion, peripheral edema, headache, flushing, and nausea and did not appear to be dose related. Both FDA approved endothelin antagonists bosentan and ambrisentan are pregnancy category X. Thus, it is our practice to recommend two forms of birth control in patients who are not sterile. Liver enzymes should be monitored with bosentan or sitaxsentan. Decreases in hemoglobin concentration of around 0.8 g/dL have been observed and are recognized as a class effect, but not dose dependent. 29 Bosentan is approximately $55,890 and ambrisentan $56,736 a year. 9
Phosphodiesterase Inhibitors (PDE5-I)
Nitric oxide (NO) is a labile gas that can diffuse across cell membranes. NO increases intracellular calcium through cyclic GMP leading to vascular smooth muscle relaxation. Cyclic GMP is degraded by phosphodiesterase type 5 in both the lung and corpus cavernosum. Inhibition of phosphodiesterase type 5 causes vasodilatation of the pulmonary arteries. NO is synthesized by endothelial NO synthase, and NO synthase deficient mice develop pulmonary hypertension. 31 Sildenafil is an orally active PDE5 inhibitor approved for the treatment of erectile dysfunction. It acutely reduces pulmonary artery pressure in patients with PAH and may alter cardiac function and vascular remodeling in the long-term.32–34 In a double-blind placebo-controlled study (SUPER-1), 278 patients with PAH were randomly assigned to placebo or sildenafil (20, 40, or 80 mg). The 6-MWD, WHO functional class, and mPAP improved for all doses of sildenafil. 222 patients completed one year of treatment with sildenafil monotherapy and had improvement from baseline in the 6-MWD of 51 m. Side effects include headache flushing, epistaxis, dyspepsia, and diarrhea. 35 Tadalafil, a longer-acting phosphodiesterase inhibitor was studied in 405 patients who were either treatment-naïve or receiving bosentan therapy. Tadalafil 40 mg was well tolerated and improved 6-MWD, quality of life, and reduced clinical worsening. Side effects were headache, myalgias, and flushing. 36 The longer half-life, 17.5 hours, of tadalafil allows once-daily dosing. Phosphodiesterase type-5 inhibitors potentiate the hypotensive effects of antihypertensive agents and are contraindicated in patients using organic nitrates. Both sildenafil and tadalafil have received FDA approval for PAH. The approximate annual cost for sildenafil is $12,761 and tadalafil will cost similarly to sildenafil. 9
Calcium Channel Blockers (CCB)
Calcium channel antagonists were the first agents to be used systematically in PAH. 37 CCBs decrease PVR by inhibiting calcium influx into the pulmonary vasculature hence leading to vasodilatation. It has been widely recognized that only a small minority of patients (< 10%) responded to treatment with CCBs. 38 Modern research has established appropriate criteria to identify patients likely to benefit from CCB treatment. With a pulmonary artery catheter in place, patients with idiopathic and HPAH should undergo an acute vasodilator challenge with either inhaled nitric oxide or intravenous adenosine. In our practice, we perform vasodilator challenges in all patients being evaluated for PAH. A positive response is defined by a decrease in mPAP by at least 10 mmHg to 40 mmHg or less without decreasing cardiac output. 39 These patients generally qualify for calcium channel antagonists therapy and have a more favorable prognosis. In 2005, a retrospective study preformed in 557 IPAH patients demonstrated vasoreactivity in only 12.6% of the patients, and only 6.8% had long-term improvement on CCB therapy defined as achieving NYHA functional class I or II with near-normal hemodynamics for at least one year with treatment. The patients with longterm benefit were also the subgroup of patients that demonstrated a more pronounced decrease in both mPAP and PVR. 38 Based on the results from this clinical study, CCB are indicated in a minority of patients with PAH. It is important to observe that oral CCB may be harmful if given to non-responders by decreasing cardiac output and systemic vascular resistance. In addition, acute vasodilator testing and chronic treatment with calcium channel blockers is contraindicated in patients with severe right heart failure. Long-acting nifedipine, diltiazem, or amlodipine are suggested and generally administered at higher doses that would be otherwise used for systemic hypertension, e.g. nifedipine XL 90 mg twice daily. Verapamil should be avoided secondary to potential negative inotropic effects. Dose escalation should be preformed if patients continue to have a vasodilator response on repeat cardiac catheterization, or if patients remain symptomatic and are not limited by hypotension or bradycardia. CCB therapy is generally well tolerated with the exception of leg edema, though this can usually be managed with compression stockings or occasionally low dose diuretics. In summary, a minority of patients are acute vasodilator responders using strict criteria, however it is critical to identify these patients and treat them with CCBs as they have a very favorable prognosis.
Combination Therapy
Although multiple randomized controlled trials of combination therapy in PAH are currently ongoing, several small combination trials have been published. The addition of bosentan to intravenous epoprostenol did not provide significant benefit. 40 There have been conflicting results in patients currently on bosentan therapy who were randomized to inhaled iloprost or placebo.41,42 Patients on intravenous epoprostenol randomized to sildenafil or placebo appear to have significant improvement in 6-MWD. 43 Further investigation into combination therapy for PAH is necessary.
Place in Therapy
Treatment for PAH is complex and individualized, and should be prescribed by specialists trained in pulmonary vascular disease. Prior to initiating therapy, other causes of pulmonary hypertension such as pulmonary venous hypertension, chronic lung disease with hypoxia, and pulmonary thromboembolic disease must be ruled-out. A right heart catheterization is essential in confirming the diagnosis, and patients with true vasodilator response should receive oral calcium channel blockers. Therapy should take into account patient's preference, severity of disease (poor prognostic indicators include evidence of right heart failure on exam, right heart dysfunction on echocardiogram, decreased CI, increased right atrial pressure, and elevated BNP), and cost. Less severe disease (class II and possibly class III) may be initially treated with oral drugs such as endothelin antagonists or phosphodiesterase-5 inhibitors. However, close follow-up is essential to determine response to therapy. If patients have not improved with oral therapy and remain function class II or have unfavorable prognostic findings, intravenous therapy should be considered. In our practice patients with advanced class III-IV functional class are usually initiated on intravenous therapy. We generally initiate epoprostenol since it offers a cost savings over the others-a generic brand is available. Patients receiving epoprostenol who remain in NYHA class III-IV despite treatment should be referred for lung transplantation. Lastly, practitioners are encouraged to enroll patients in clinical trials.
Patient Preference
For many patients, oral medications have the highest patient satisfaction since they are easy to administer and have rare serious side effects. However, they are not appropriate for all patients, and patients with advanced PAH are unlikely to respond to oral drugs alone. For example, oral therapy is not first-line treatment in patients with class IV PAH, and many patients feel poorly enough that they are willing to tolerate the prostaglandin side effects. Patients who are not willing to have an indwelling catheter are clearly not candidates for IV epoprostenol or IV treprostinil. In our experience, almost all patients using subcutaneous treprostinil develop site pain and erythema; hence patients usually prefer IV administration to subcutaneous therapy. Because of survival and cost advantages as well as historical experience, we generally use epoprostenol IV in patients with advanced PAH particularly in those with substantial heart failure. Inhaled iloprost is dosed six times a day which is difficult for most patients; hopefully, with the recent approval of inhaled treprostinil, there will be better adherence to inhaled treatment. Lastly, PAH medications are very costly, and patient finances may limit therapeutic options. Several patient assistance programs are available to cover the cost of PAH drugs, and experienced pulmonary hypertension centers have resources to help patients through these assistance programs.
Future Directions
Imatinib mesylate is a protein-tyrosine kinase inhibitor that inhibits Bcr-Abl, platelet-derived growth factor, stem cell factor, and c-kit. Imatinib has been studied in human trials in patients failing active PAH treatment. Patients were randomized to 200 mg imatinib daily (increased to 400 mg daily as tolerated) or placebo. The primary end-point, improvement in 6-MWD, was not achieved; however, there was significant improvement in hemodynamic parameters. 44 A future trial in patients with severe PAH, IMPRES, is currently planned. 20
Rho/Rho-kinase signaling is believed to play an important role in mediating acute pulmonary vasoconstriction and vascular remodeling in PAH. In previous studies, Rho/Rho-kinase inhibitors have been shown to reduce pulmonary artery pressure in monocrotaline-induced PAH and in chronically hypoxic rats.45,46 In our lab, we demonstrated that chronic oral fasudil administered to
Other potential therapies currently being investigated include PPRgamma agonists, angiotensin converting enzyme 2, selective serotonin receptor inhibitors, vasoactive intestinal peptide, soluble glucocorticoid stimulators, tetrahydrobiopterin, aspirin, statins, and serine protease inhibitors.20,48–54 Sex hormone manipulation is another potential therapeutic target given the female predominance in PAH. 55 Finally, since right ventricular function is the primary determinant of survival in PAH, future therapies may directly target the right ventricle. 56
Conclusions
There have been exciting pharmacologic advancements in the field of pulmonary arterial hypertension in the past several decades. This article has reviewed the therapeutic advances in PAH including prostacyclin analogues, endothelin receptor antagonists, and phosphodiesterase-5 inhibitors. These therapies have improved symptoms, hemodynamics, and even survival; yet, PAH continues to remain a progressive, incurable disease. Researchers are actively investigating combination therapy and new classes of drug therapy. We hope to see several new classes of PAH therapy in the coming years, and with these new therapies transform PAH from a treatable disease to a curable one.
Disclosures
The authors report no conflicts of interest.