
Abstract
Sickle cell disease is caused due to a genetic disorder, which accounts for people dying at an early age in Nigeria. A retrospective study of sickle cell disease patients was carried out with a view to determining the disease pattern in sickle cell patients from the Northwestern Nigeria. Case notes of 319 sickle cell patients were collected and reviewed retrospectively. The prevalence of sickle cell trait, comorbidity of sickle cell disease and malaria, and the effects of sickle cell disease and age on the weight and hematological parameters of sickle cell patients were determined and analyzed. Results showed the prevalence rate of sickle cell trait to be 61.8% (197) and that of non-sickle cell trait to be 38.2% (122). The sickle cell trait comprised 96 males (48.7%) and 101 females (51.3%). Among these patients, 51 (41.8%) males and 71 (58.2%) females had malaria. However, 35.4% (113) of sickle cell patients and 7.5% (24) of malaria patients showed anemia. Genotyping revealed 32 AS (16.2%), 102 SS (51.8%), SS+F (3.6%), and 56 SC (28.4%). The associated prevalence rates of clinical signs were pain/crisis 45.1% (89), pneumonia 28.4% (56), gastric disorders 9.1% (18), central nervous system (CNS) disorders 4.1% (8), renal diseases 2.5% (5), musculo-skeletal disorders 2.5% (5), conjunctivitis 0.5% (1), acute chest syndrome 0.5% (1), cholecystitis 0.5% (1), hemophilia 0.5% (1), fever 0.5% (1), priapism 2.0% (4), splenomegaly 2.0% (4), and epistaxis 1.5% (3). Few patients lived up to 49 years. There was significant difference (P < 0.05) in hematological parameters of the patients from various age groups. The use of anti-sickling, hematonic, analgesic, anti-inflammatory, and antimalarial drugs in the treatment of the affected disease in patients might have improved their quality of life.
Introduction
Protection against some infectious diseases from inheritance of polymorphism has been established. Malaria is the most significant disease affecting the expression of blood groups. Abnormal red blood cells such as Fy (a-b-) phenotype and the S-S- phenotypes have been frequently observed in Africa and South East Asia. 1 Sickle cell anemia patients between the age of two and four years are prone to greatest risk of developing anemic crisis. 2 In the United States, the life expectancy of sickle cell patients is reduced by about 30 years. 3 Sickle cell disease (SCD), an inherited disorder of hemoglobin, occurs in 70,000 to 80,000 Americans of African, Mediterranean, or Middle Eastern origin. 4 Nigeria has the largest population of people living with sickle cell disorder, with about 150,000 births annually. 5 More than 300,000 babies are born worldwide with SCD mostly in low- and middle-income countries, with the majority of these births in Africa. 6 In Nigeria, a quarter of the affected children are diagnosed before infancy and three-quarters before they complete three years of age. 7 The prevalence of sickle cell anemia ranges from 2 to 3% of the population. 8 Sickle cell anemia causes low mean weight and height. 9 The risk of asymptomatic bacterium is three times more common in Nigerian children with sickle cell anemia than in children with normal hemoglobin count. 10 Children with sickle cell anemia are more prone to developing urinary tract infections (UTIs) and other bacterial infections than those with normal levels of hemoglobin 11 and may have compromised kidney function from repeated vaso-occlusive episodes and recurrent UTI. 12 The disease is also associated with retinopathy, with higher prevalence among S-S men than among SS women. 13 The organisms causing bacteremia in African children with sickle cell anemia are the same as those in developed countries. Therefore, the use of conjugate vaccine against Streptococcus pneumoniae and Haemophilus influenzae can improve the quality of life of the patients. 14 Complications of SCD occurred in 25% of pregnancies, with birth weight of infants being below 2.5 kg in 20% of pregnancies. 15 In Nigeria, the majority of patients with sickle cell anemia are 10 years and below 16 as opposed to 18 to 43 years in the United Kingdom. 15 Sickle cell anemia is responsible for death of 25% of children under the age of five years in Africa. 17 The three most common musculoskeletal complications seen in older patients with higher platelets count are leg ulcer, avascular necrosis, and osteomyelitis. 18 High hemoglobin levels appear to be an important factor for painful crisis. 19 In Sudan, 54% of target samples were heterezygous carrier (HbAS), 42% were normal (HbAA), and 4% were diagnosed with SCD. 20 The only SCD-modifying drug is hydroxyurea. But the new Aesloz works via binding to hemoglobin and produces antisickling effect in red blood cells. 21 In view of the serious health implications of sickle cell anemia, the comorbidity of malaria in SCD was retrospectively studied among patients presented to the Department of Haematology, Usmanu Danfodiyo University, Sokoto, Northwestern Nigeria.
Materials and Methods
Case notes (319) of patients presented at the Haematology Department, Usmanu Danfodiyo University Teaching Hospital, Northwestern Nigeria, were collected and reviewed retrospectively. The review period was between January 1999 and December 2013. Data were sorted out according to age, sex, weight, and incriminating hematological diseases. Cases of sickle cell anemia and malaria were sorted out from other hematological cases. Sickle cell disorders were also classified based on genotype. The method of Saganuwan and Onyeyili, 22 Ganong, 23 and Guyton and Hall 24 was used to determine the total blood volume of patients using 8% of their body weights. The total blood volume was determined by multiplying the plasma volume by 100/100 minus hematocrit, and red cell volume was determined by subtracting the plasma volume from the total blood volume. Microscopy and electrophoresis of red blood cells were used for the diagnosis of malaria and SCD respectively. In addition, therapeutic regimens of SCD and malaria presented to the hospital were reviewed. All procedures were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the 1975 Declaration of Helsinki, as revised in 2008. Informed consent was obtained from all the patients for being included in the study.
Data analysis
Data on sickle cell and malaria were analyzed using chi-square at 0.1% level of significance. However, data on incidence of sickle cell traits were presented in percentage and their mean hemoglobin values were determined. Data on weight, packed cell volume, total blood volume, red cell volume, plasma volume, and hemoglobin were analyzed using analysis of variance (ANOVA) and the least significant difference was detected at 5% level.25,26
Results
Table 1 presents the epidemiology of SCD presented to the Haematology Department of Usmanu Danfodiyo University Teaching Hospital within the study period (2009–2013). Out of 319 patients presented to the hospital, 197 (61.8%) were diagnosed with sickle cell trait, comprising 96 (48.7%) males and 101 (51.3%) females respectively. But a total of 122 (38.2%) patients were diagnosed with malaria. A total of 147 (46.1%) male and 172 (53.9%) females were presented to the hospital (Table1).
Incidence of sickle cell disease and malaria.
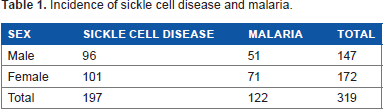
Out of 319 recorded cases, there were 113 (57.4%) cases of anemia due to SCD and 24 (19.7%) caused by malaria. But 84 (42.6%) and 98 (80.3%) neither showed anemia due to malaria nor sickle cell trait (Table 2).
Incidences of anemia caused by sickle cell and malaria.
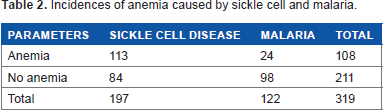
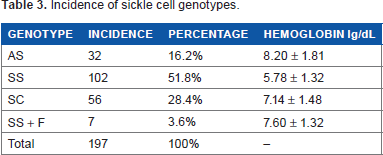
Table 3 shows the incidence of genotypes of sickle cell hemoglobin. Out of 197 recorded genotypes, 32 (16.2%) belonged to the genotype HbA, HbS, whereas 102 (51.8%) belonged to the HbS HbS, 56 (28.4%) belonged to HbS HbC, and 7 (3.6%) belonged to Hbs Hbs + F respectively (Table 3).
Incidence of sickle cell genotypes.
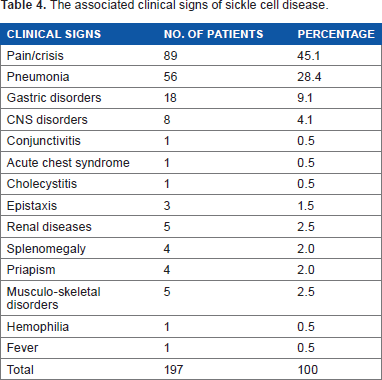
The observed clinical signs and their frequencies associated with SCD are 89 (45.1%) pain/crisis, 56 (28.4%) pneumonia, 18 (9.1%) gastric disorders, 1 (0.5%) conjunctivitis, 1 (0.5%) acute chest syndrome, 1 (0.5%) cholecystitis, 3 (1.5%) epistaxis, 5 (2.5%) renal diseases, 4 (2.0%) splenomegaly, 4 (2.0%) priapism, 5 (2.5%) musculoskeletal diseases, 1 (0.5%) hemophilia, and 1 (0.5%) fever. Pneumonia was caused by Streptococcus pneumonia (Table 4). The observed disorders related to CNS are ischemic heart disease, hypertension, and sleeplessness. However, the observed renal diseases are anuria, urinary tract infection, and polycystitis. But the musculo-skeletal-related abnormalities observed are avascular necrosis of femur head, chronic osteomyelitis of right tibia, arthritis, and acute osteomyelitis. But the gastrointestinal disorders observed are gastritis, gastroparesis, and atrophic gastritis.
The associated clinical signs of sickle cell disease.
The bodyweight of sickle cell patients within the age ranges of 10–14 years, 15–19 years, 20–24 years, 25–29 years, 35–39 years, and 45–49 years were significantly higher (P < 0.05) than the weight of the patients within the age ranges of 0–4 years and 5–9 years. But there was no significant difference (P > 0.05) in the packed cell volume of all the sickle patients except for those within the age range 45–49 years, which was significantly decreased (P < 0.05). But their range of packed cell volume was between 18.00 ± 0.00% and 21.71 ± 2.62%. However, patients within the age ranges of 15–19 years, 20–24 years, 25–29 years, and 35–39 years recorded significantly (P < 0.05) increased the total blood volume of 3.15 ± 0.13 L, 3.16 ± 0.64 L, 4.36 ± 0.96 L, and 4.10 ± 0.00 L when compared to patients within the age ranges of 0–4 years (1.48 ± 0.38 L), 5–9 years (1.6 ± 0.22 L), and 45–49 years (1.92 ± 0.00 L) respectively. However, red blood cells volume of patients within the age ranges of 15–19 years (0.60 ± 0.01 L) and 20–24 years (0.67 ± 0.02 L) were significantly higher than the red cells volumes of the patients within the age range 0–4 years (0.32 ± 0.02 L), 5–9 years (0.37 ± 0.01), 10–14 years (0.46 ± 0.01), and 45–49 years (0.42 ± 0.00 L), which were in turn higher than the red cells volumes of the patients within the age ranges 25–29 years (0.05 ± 0.01 L) and 35–39 years (0.04 ± 0.00 L) respectively. Plasma volume of the patients increased from 1.16 ± 0.15 L (10–14 years) to 1.30 ± 0.21 L (5–9 years), 1.93 ± 0.15 L (10–14 years), 2.55 ± 0.12 L (15–19 years), 2.49 ± 0.65 L (20–24 years), 3.50 ± 0.95 L (25–29 years), and 4.06 ± 0.00 L (35–39 years), respectively. But in the age range of 45–49, the plasma volume (1.57 ± 0.00 L) decreased significantly (P < 0.05). The patients within the age ranges 0–4 years (6.20 ± 1.61 L), 5–9 years (6.95 ± 0.95 L), 10–14 years (9.97 ± 0.41 years), 25–29 years (6.33 ± 0.33 L), 35–39 years (6.33 ± 0.00 L), and 45–49 years (6.00 ± 0.00 L) recorded significantly decreased (P < 0.05) hemoglobin as compared with the patients within the age ranges 10–14 years (9.97 ± 0.61 L), 15–19 years (13.11 ± 2.67 L), and 20–24 years (13.15 ± 1.70 L), respectively (Table 5). However, in our study, there were no recorded cases of patients within the age ranges 30–34 years and 40–44 years.
Age, weight, and hematological parameters of sickle cell disease patients.
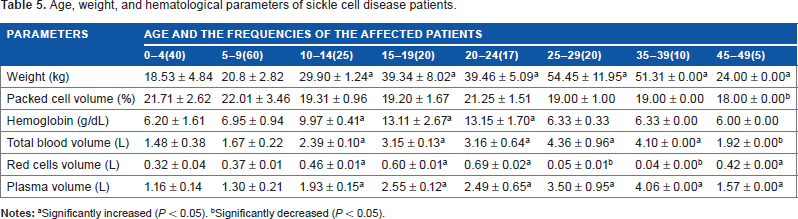
Significantly increased (P < 0.05).
Significantly decreased (P < 0.05).
Discussion
SCD is prevalent in many parts of the world, especially among Nigerians and Americans of African, Mediterranean, and Middle East origin. The fact that 61.8% (197) of the patients were diagnosed with sickle cell trait and 38.2% (122) were diagnosed with malaria is an indication that 6 in every 10 cases recorded in the hospital are of sickle cell origin. Our findings disagree with the reports of WHO 27 and Hoffbrand et al. 28 indicating that West African countries including Nigeria has the frequency of the trait ranging from 15 to 50%. In the present study, females (51.3%) are more affected than males (48.7%). Baum et al. 19 reported that there was striking increase in painful crisis in male patients. But the remaining 38.2% of patients diagnosed of malaria indicates that malaria constitutes significant problem in the northwestern region of Nigeria.
The reported cases of anemia caused by SCD and malaria agree with the report of George and Opara 29 indicating that anemia is a sign of malaria and SCD. Polypharmacy adopted by the hospital that involves the use of anti-sickle cell (hydroxyurea, allopurinol), antimalarial (paludrine, fansidar, coartem, lonart, artesunate), blood tonic (vitamins, B complex, C, astymin, folic acid, fesolate), analgesic (ibuprofen, paracetamol, tramol, DF118, diclofenac), and glucocorticoids (hydrocortisone, prednisolone). Surviving malaria is the most significant selective force affecting the expression of blood groups. Red cells lacking or having altered forms of blood group active molecule are commonly found in regions of the world where malaria is endemic, notably the FY (a-b) phenotype and the s-s-phenotype in Africa. 1 So the presence of SS, SS+F, and SC among the study group is corroborated by the report of Sergeant 30 indicating that SCD is caused by HbSS, HbSC, and Hb Sathal. The S gene results from the replacement of the normal codon GAG at position a^6 by GTG. As a result of this change, valine is inserted at this position instead of glutamic acid usually in that position in an HbA individual. In the deoxygenated state, the HbS molecule forms polymers, which progress to the sickle red blood cell. 30 But the 16.2% (32) prevalence rate of heterozygous (HbAS) carrier disagrees with the report of Munsoor and Alabid 20 indicating that the prevalence rate of heterozygous in Western Sudan is 54%. But in our study, 51.8% (102) were diagnosed with SCD as against 4% reported in Western Sudan. 20 Although HbAS can cause sickling symptoms during severe hypoxic states, the complications are rare. 31
The observation of pain/crisis, pneumonia, gastric, CNS, biliary, renal and musculo-skeletal disorders, acute chest syndrome, priapism, and conjunctivitis agree with the report of Isosa 32 who had earlier attributed all the above-mentioned signs to SCD. The isolation of Streptococcus pneumonia from the patients presented to the teaching hospital is corroborated by the report of Gaston et al. 33 indicating that the S. pneumonia could cause an episode of life-threatening sepsis in children suffering from SCD. The ischemic heart disease observed in our study agreed with the report of Pegelow et al. 34 and Vichinsky et al. 35 indicating that SCD can cause acute ischemic stroke in the affected adults. The acute chest syndrome (ACS) with fever observed in our study is corroborated in the report of Castro et al. 36 indicating that ACS characterized by fever and respiratory symptoms can be observed in SCD patients. A major risk factor for the development of ACS is the hemoglobin genotype. 36 ACS is the second most common cause of hospitalization in sickle cell patients. 37 But in our study, pain/crisis is the most common cause of hospitalization, followed by pneumonia, gastric disorders, renal diseases, and musculo-skeletal disorders.
Sickle hemoglobinopathy can cause visual loss in 0.5% of the affected patients. NIH 38 had earlier reported that SCD can cause conjunctivitis, which usually results in permanent, devastating loss of vision. 32 However, Cholecystitis observed in 0.5% of the sickle cell patients agrees with the report of Jawad et al. 39 indicating that cholecystitis is one of the key signs of SCD. Anuria, urinary tract infections, and polycystitis all observed in 2.5% of the sickle cell patients agree with the report of Isosa, 32 and John et al. 40 indicating that urinary tract disorders are characteristic signs of SCD.
The median survival period for patients with sickle cell anemia in the present study is 49 years and the average weight (24.00 ± 0.00 kg), PVC (18.00 ± 0.00%), Hb (6.00 ± 0.00 g/dL), TBV (1.92 ± 0.00 L), RCV (0.42 ± 0.00 L), and PV (1.57 ± 0.00 L) disagree with the report of WHO 27 indicating that there are no firm data on the median survival period of sickle cell patients from the African continent. In the United States, median survival was estimated in 1994 to be 42 years for men and 48 years for women. In Jamaica, median survival was 52 years for men and 58.5 years for women. 27 In the present study, anemia of the affected SCD patients significantly increased from the age range 0–4 years passing through 5–9 years, 25–29 years to 35–39 years with low amount of hemoglobin, total blood volume, red cell volume, and plasma volume. This may be an indication that much of the erythrocytes have been lysed. Baum et al. 19 had earlier reported that patients between the ages of 15 and 25 years experience a striking increase in painful crisis with hemoglobin levels above 8.5 g/dL. High hemoglobin levels appear to be an important risk factor for painful crisis. The incidence of painful crisis was observed among the patients within the age group of 10–24 years with hemoglobin range 9.97 ± 0.41 to 13.15 ± 1.70 g/dL. For those patients in this age group, there was progressive anemia. Juwah et al. 2 had reported a gradual but progressive decline in the incidence of severe anemia in the age range 8–16 years. Management of pain associated with SCD consists of the use of non-steroidal anti-inflammatory drugs (NSAIDs), opioids, and adjuvant medications. 41 A sustained release opioid preparation provides more consistent analgesia 38 and by so doing facilitate rest. 32 The significantly increased total blood volume, red cells volume, and plasma volume of 15–39 years may be due to increased administration of blood tonic, allopurinol, and hydroxyurea. Frenette and Atweh 42 had earlier reported that hydroxyurea can induce HbF production in SCD. Hydroxyurea is an S phase-specific chemotherapeutic agent that caused a marked increase in Hb F levels in baboons, 43 decrease in the frequency of painful crisis, acute chest syndrome, reduction in transfusion requirements, and hospitalization in adults with moderate to severe SCD 44 invariably causing improved survival rate. 45 Hydroxyurea reduces circulating white blood cell count and perhaps also the number of adherent leukocytes recruited to the wall of small venules, which was correlated with the clinical hydroxyurea.46–48
Conclusion
The prevalence rate of SCD in the northwestern Nigeria is 61.8%. The affected individuals showed significant loss of body weight, blood, and sickle cell pain/crisis and can survive up to the age of 49 years. The use of anti-sickling, hematonic, analgesic, anti-inflammatory, and antimalarial drugs in the treatment of the affected patients might have improved the quality of life of the affected individuals.
Author contributions
SAS generated and interpreted the data and wrote the manuscript. The author reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
I sincerely thank Dr. Alhaji Muhammad Ndakotsu, Head of the Department of Pathology, College of Health Sciences, Usmanu Danfodiyo University, Sokoto for attending to the patients and allowing me to collect the data from the Department. He also provided an enabling environment during the period of data collection.
I also thank the entire management and technologists in the Departments of Haematology, Microbiology, Radiology, Medicine and Ophthalmology, Usmanu Danfodiyo University, Sokoto Teaching Hospital, for their contributions in various capacities. The contribution of Kehinde Ola Emmanuel (who typed the work), National Open University, Nigeria, is highly appreciable.