
Abstract
A new era in cancer therapy is emerging with the development of tumor-specific agents exhibiting less toxicity. Since the advent of imatinib, several tumor-directed treatment options have been developed. However, therapies not directed specifically at a tumor target also have potential benefits. The 26S proteasome is a critical regulator of cell homeostasis through the degradation of key signaling molecules including p21, p27, and p53. Additionally, the proteasome degrades I-κB which inhibits the activity of NF-κB, an important promoter of cell proliferation. Blocking function of the proteasome disrupts tumor growth by shifting the balance of the cell from proliferation to apoptosis. In vitro, the proteasome inhibitor, bortezomib, inhibits NF-κB activity and prevents growth of several malignant cell types including multiple myeloma. Given the central role of NF-κB in the pathogenesis of multiple myeloma, bortezomib was a good candidate for use in therapy. Treatment of heavily pre-treated patients with bortezomib led to response rates of 30%-40%. More importantly, bortezomib led to improvements in bone metabolism, a major cause of morbidity in multiple myeloma. This effect was seen independent of the response of the myeloma. This finding correlates with in vitro studies which demonstrate increased BMP2 expression and osteoblast number after exposure to bortezomib. Moreover, bortezomib blocks NF-κB-mediated angiogenesis and tumor cell metastasis. While tumor-targeted treatments have an important role in the future of cancer therapy, these examples show that it is important not to lose sight of the benefits of less-specific agents in the treatment of malignant neoplasms.
Introduction
Numerous advances in the treatment of cancer have been made over the last five decades. Progressing from radiation to single agent therapy to complex multidrug regimens has improved the outcomes of patients with many types of cancer. However, conventional cancer chemotherapy has its drawbacks. Traditional therapeutic agents were developed based on their abilities to kill rapidly dividing cells. However, these agents are not selective in their destructive properties and can lead to acute toxicities, organ damage, and even second malignancies. These realities of cancer chemotherapy have spurred the development of more tumor-specific therapies. The first success story, imatinib (Gleevec®), has revolutionized the treatment of chronic myelogenous leukemia (CML). Directed at the hybrid enzyme protein created by the BCR-ABL gene fusion, imatinib is able to selectively target CML cells while limiting damage to normal tissues. 1 Prior to the use of imatinib, many patients with CML did not respond to front-line therapies and the outcomes were suboptimal with survival rates at about 70%.2,3 Incorporating imatinib into current CML therapy regimens has improved outcomes in these patients with minimal increase in toxicity. Data from the International Randomized Interferon Study (IRIS) reveals a 5-year survival rate of 92% with complete hematologic responses achieved in 96% of patients who received imatinib. 4 Since the advent of imatinib, multiple novel therapeutic options have emerged including directed therapy at tumor cell proteins (gemtuzumab, rituximab), tumor vasculature (bevacizumab), and cellular growth (trastuzumab).5–8 Each of these agents has had varying degrees of success in improving the outcomes of patients with a wide range of malignancies, and in general, they have exhibited less toxicity than traditional chemotherapeutic drugs. It is likely that continued understanding of cancer cell pathophysiology and advances in drug development will result in the creation of even more precise instruments of cancer destruction and hopefully provide the magic bullets oncologists seek.
While searching for the holy grail of tumor-specific therapy is certainly a worthwhile endeavor, there might actually be advantages to agents that have a less precise approach and their applications in the treatment of human malignancies. In the current issue of Clinical Medicine: Therapeutics, Dr. G. David Roodman describes advances in the treatment of multiple myeloma with a focus on the use of the proteasome inhibitor, bortezomib. Proteasome inhibitors represent a new class of therapeutic agents directed towards a novel target. Instead of targeting cell division machinery or DNA replication like traditional chemotherapy agents, proteasome inhibitors alter the physiologic balance of the cell by preventing the degradation of key regulatory molecules.
Proteasome Inhibition and Multiple Myeloma
The 26S proteasome is a large multi-subunit complex critical for maintaining cell homeostasis through the destruction of cell signaling and regulatory proteins. It is composed of three major components: two 19S regulatory subunits and the 20S core which contains the proteolytic function (Fig. 1). 9 The 20S core is comprised of 4 heptameric subunits, two α and two β. The β-subunits contain the proteolytic activity including a tryptic site, chymotryptic site, and a peptidylglutamyl site (Fig. 1). 9 Proteins that are marked for degradation are poly-ubiquitinated through the coordinated actions of ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3) with the E3 ligase providing specificity in protein targeting. 10 Poly-ubiquitinated proteins are then shuttled through the 19S regulatory subunit of the proteasome into the proteolytic 20S core where they are degraded. 10 Bortezomib inhibits the 26S proteasome by reversibly binding to the β5 subunit of the 20S core and inhibiting its chymotrypsin-like activity, thus disabling the ability of the proteasome to break down proteins and preventing further protein entry into the proteasome.11,12
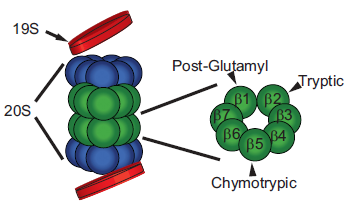
Structure of the 26S proteasome. The 26S proteasome is comprised of two 19S regulatory subunits and a 20S proteolytic core (left). The 20s core is made up of 2 α-subunits (blue) and 2 β-subunits (green). Close up of the β-subunit reveals the sites of proteolytic activity (right). Bortezomib binds the b5 subunit and inhibits the chymotryptic activity. Adapted from 9 .
In his article, Dr. Roodman elegantly addresses the therapeutic modalities available for the treatment of multiple myeloma with a focus on bortezomib. NF-κB was the initial signaling molecule focused on in the study of proteasome inhibition in myeloma. It plays a central role in the regulation of many cell functions including cell growth and differentiation, cytokine and growth factor secretion, immune and inflammatory responses, and angiogenesis. 13 The activity of NF-κB is regulated by its inhibitory protein, I-κB, which forms a stable complex with NF-κB and prevents its translocation into the nucleus to regulate transcription.13,14 When activated by an enzyme such as I-κB kinase (IKK), I-κB is phosphorylated and then ubiquitinated and shuttled to the proteasome for degradation. Upon its release, NF-κB translocates into the nucleus and activates transcription. Bortezomib interferes with this process by blocking the degradation of I-κB leading to increased levels which can sequester NF-κB in the cytoplasm and prevent activation of transcription.
NF-κB plays an important role in the pathogenesis of multiple myeloma. Myeloma cells isolated from patients demonstrate constitutive NF-κB activity which promotes proliferation, cell survival, and protection from chemotherapy-induced apoptosis. 15 Increased NF-κB activity has been linked with treatment resistance. 16 Perhaps more importantly, the bone marrow microenvironment promotes tumor growth via NF-κB-dependent processes. Upon binding to marrow stromal cells, myeloma cells activate multiple signaling pathways, including NF-κB, through interactions between adhesive cell surface proteins, such as cadherin and integrins.17,18 This interaction leads to the secretion of multiple growth factors including VEGF, IL-6, TNF-α, IGF-1, and RANK ligands. 15 NF-κB-mediated IL-6 secretion is also protective against myeloma cell death. 19 These examples demonstrate how both internal and external factors play as crucial a role in the growth and survival of malignant cells. Bortezomib functions by disrupting the cross-talk between the myeloma cells and the bone marrow stroma. Treatment of myeloma cells with bortezomib inhibits NF-κB-mediated IL-6 production, an important growth and anti-apoptotic factor. 17 Furthermore, proteasome inhibition prevents adherence of multiple myeloma cells to bone marrow stroma, cutting off the myeloma cell's support system. 17 Additionally, the in vitro activity of bortezomib has been confirmed in in vivo models. Bortezomib has activity in mouse xenografts 20 and sensitizes myeloma cells to other therapeutic agents. 17
Given the very promising pre-clinical data, bortezomib entered the clinical arena. A phase I trial in patients with recurrent or refractory malignancies showed promising results, but more importantly, the drug was well tolerated with thrombocytopenia being the prominent adverse effect. 21 These encouraging results along with the favorable toxicity profile propelled bortezomib in to phase II testing for multiple myeloma. Once again, bortezomib showed beneficial effects, even in patients who had been heavily pretreated. (Table 1)22,23 Furthermore, in addition to eradicating the myeloma cells, patients taking bortezomib showed signs of improved bone metabolism. Patients with multiple myeloma demonstrated increased levels of bone-specific alkaline phosphatase and osteocalcin indicating augmented osteoblast activity. 24 This effect is not seen in patients receiving other myeloma therapies and is present irrespective of the response of the patient's myeloma indicating that this effect is specific for bortezomib on bone formation. 25 This demonstrates how less tumor-specific therapies can lead to a more global benefit to the patient.
Selected single-agent bortezomib trials in hematologic malignancies and solid tumors.
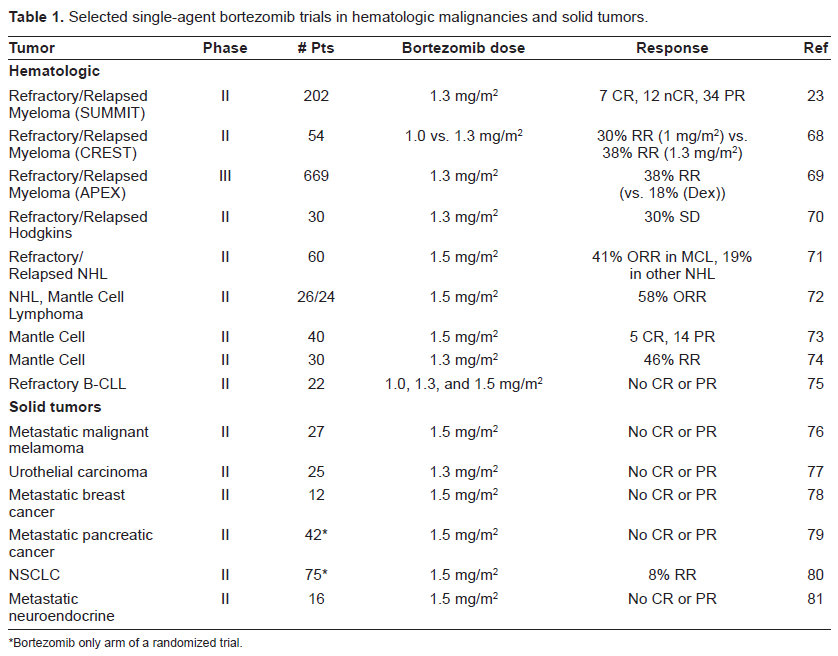
Bortezomib only arm of a randomized trial.
Use of Proteasome Inhibitors in other Malignancies
Although multiple myeloma has been the initial focus of proteasome inhibition, drugs like bortezomib have applications in several other cancers. NF-κB has been implicated in the etiology and treatment response of various other malignancies. Like in multiple myeloma, NF-κB is constitutively active in several other cancers including melanoma as well as in breast, ovarian, thyroid, colon, and prostate cancers.26–32 Furthermore, NF-κB is often activated by treatment which limits the efficacy of drugs against the tumors.33–35 Clinically, increased NF-κB activity has been linked with treatment resistance in multiple myeloma. 16 NF-κB mediates chemoresistance through upregulation of anti-apoptotic factors such as cellular inhibitors of apoptosis (Bcl-xL, cIAP1, cIAP2, XIAP), the Bcl-2 homologue, A1/Bfl-1, and the A20 zinc finger protein.36–39 In squamous cell carcinoma, bortezomib treatment was accompanied by decreased NF-κB-dependent cytokine and growth factor secretion and angiogenesis which in turn inhibited cancer cell proliferation. 40 Given the potential benefits of bortezomib in several cancer models, multiple trials have been conducted to explore the clinical efficacy of bortezomib in various other malignancies. Details of selected trials of bortezomib as a single agent are summarized in Table 1.
The effects of proteasome inhibitors extend beyond the NF-κB pathway. p21 and p27, which are important in the control of cell cycle and replication, are also regulated by the proteasome.41,42 In colorectal and breast cancer, low levels of p27 are correlated with a poor prognosis.43,44 Additionally, the proteasome also controls levels of the pro-apoptotic transcription factor, p53. Regulated by MDM2 which targets it for proteasomal degradation, p53 is frequently found at low levels in cancer cells; 45 treatment of fibroblasts and pheochromocytoma cells with bortezomib leads to accumulation of p53.46,47 Pro-apoptotic molecules such as Bax, Bim, and Bid, which inhibit the activity of the anti-apoptotic Bcl-2 protein family members and inhibitors of apoptosis (IAP), are also regulated by proteasomal degradation.48–50 In addition to modulating cell signaling, proteasome inhibition as also able to induce apoptosis as a part of the unfolded protein response (UPR) (for review see 51 ). Briefly, the unfolded protein response is a protective mechanism to prevent overloading of the endoplasmic reticulum (ER) with improperly folded proteins in the face of environmental stresses. The first phase of this response involves multiple processes including a slowing of translation to decrease the protein load in the ER, upregulation of ER chaperone proteins and folding enzymes, and activation of the ER associated degradation (ERAD) apparatus which translocates misfolded proteins to the cytoplasm and into the 26S proteasome for degradation. In cases where the UPR is inadequate to relieve the backup of unfolded proteins, apoptotic pathways are activated leading to death of the cell. Blockade of protein breakdown, a critical component of the UPR, via proteasome inhibition promotes failure of the UPR and pushes the cell towards apoptosis. This pathway is especially important in both normal plasma and multiple myeloma cells. These cells produce large quantities of immunoglobulins and have high throughput through the ER. 52 Disruption of the UPR in these cells leads to rapid accumulation of unfolded proteins and activation of apoptosis.53,54 This is perhaps a major component of the unique sensitivity of multiple myeloma as well as normal plasma cells, to proteasome inhibition. This may also explain the better response of hematologic malignancies to bortezomib than seen in solid tumor trials (Table 1).
Effect of Proteasome Inhibition on the Tumor Microenvironment
Much of the early work on proteasome inhibitors has focused on what they do to the physiology within the tumor cell. We are now discovering that the effect of proteasome inhibition extends beyond the tumor cell wall. As mentioned above, NF-κB plays a central role in cell proliferation, but also in the control of cytokine and growth factor secretion. The interactions of myeloma cells and the bone marrow stroma discussed above is a good example. As mentioned above, in addition to disrupting the relationship between the myeloma cells and the marrow stroma, bortezomib has beneficial effects on the bone loss seen in myeloma, a major source of morbidity in this disease. Treatment of a myeloma mouse model with bortezomib reveals increased bone mineral density. 55 Furthermore, increased numbers of osteoblasts were noted with an accompanying decrease in osteoclast number indicating a shift towards bone formation. Also, proteasome inhibition led to increased BMP2 expression, an inducer of osteoblast differentiation and bone formation. 56 Similar results have been seen in human osteoblast precursor cultures from patients with myeloma. 57 Modulation of angiogenesis can also have a significant impact on tumor survival. In squamous cell carcinoma, treatment with proteasome inhibitors prevents the expression of pro-angiogenic cytokines, vascular endothelial growth factor, and growth-related oncogene-α leading to decreased vascularity. 40 Finally, NF-κB modulates factors involved in tumor cell metastasis. Cell adhesion molecules such as E-selectin, ICAM-1, and VCAM-1 are regulated by NF-κB. 58 In mice injected with Lewis lung carcinoma cells, bortezomib reduced metastatic disease. 59 These examples demonstrate how proteasome inhibitors can modify the tumor microenvironment and surrounding tissue in addition to the direct cytotoxic effects on the tumor cells themselves.
In addition to the efficacy in controlling tumors and their microenvironments, the broad effects of proteasome inhibition can be useful in the treatment of more heterogeneous tumors. Neuroblastoma, a common childhood tumor, is comprised of three main cell types: neuroblastic (N-type), stromal (S-type), and intermediate (I-type) which have features of both N- and S-type cells.60,61 The N-type cells comprise the bulk of the neuroblastoma tumors and are thought to be the main malignant component. 60 However, the S-type cells also appear to play a critical role in tumor survival and can repopulate the tumor even after elimination of the N-type cells. 62 Interestingly, N-type cells have low baseline NF-κB activity and treatment of N-type cells with doxorubicin or etoposide lead to apoptosis through the activation of NF-κB. 63 Conversely, S-type cells have high basal NF-κB activity and inhibition of that activity leads to cell death. 64 These contrasting cell physiologies pose a problem to the development of specific, targeted therapies to neuroblastoma. However, despite these differences, bortezomib is cytotoxic to all three cell subtypes as a single agent. When used in combination with standard chemotherapy agents, bortezomib sensitizes the cells to the second agent, often leading to synergistic effects. 65 Likely, the disturbance of the overall neuroblastoma cell homeostasis leads to destabilization of the cell and sensitization to chemotherapy. Even in a chemoresistant neuroblastoma cell line, SK-N-BE(2), which has a mutation in p53 rendering it non-functional, 66 bortezomib is active as a single agent and addition of bortezomib to the other chemotherapy drugs sensitizes the cells to the second agent. 65 This example demonstrates the benefits that a non-tumor specific drug like bortezomib can have in neoplastic control of complex tumors where directed therapy alone is likely going to be ineffective.
Conclusion
Our knowledge of the mechanisms of action and the impact of proteasome inhibition continues to increase. We are not only elucidating the specific pathways involved, but also understanding the broader context of the effects seen with treatment. Furthermore, newer, more potent proteasome inhibitors are in development and testing. 67 What is important now is to continue thinking outside the box and understanding the impact of proteasome inhibition on not only the tumors, but also on the cells and tissue surrounding them. By examining this in addition to what is occurring inside the cancer cell, we may be able to more optimally apply this class of drugs in the treatment of various malignancies. Furthermore, more effective therapeutic combinations can be developed to improve the outcomes of patients with complex diseases such as neuroblastoma and multiple myeloma. While the development of tumor-directed therapies is definitely an appropriate priority, it is essential not to lose sight of the potential benefits of less specific agents in the treatment of malignant neoplasms.
Disclosure
Michael B. Armstrong M.D., Ph.D. reports no conflicts of interest.
Footnotes
Acknowledgement
The author would like to recognize Dr. Daniel Wechsler for his assistance and helpful guidance in the preparation of this manuscript