
Abstract
Fospropofol, a phosphorylated prodrug version of the popular induction agent propofol, is hydrolyzed in vivo to release active propofol, formaldehyde, and phosphate. Pharmacodynamic studies show fospropofol provides clinically useful sedation and EEG/BIS suppression while causing significantly less respiratory depression than propofol. Pain at the injection site, a common complaint with propofol, was not reported with fospropofol; the major patient complaint was transitory perianal itching during the drug's administration. Although many clinicians believe fospropofol can safely be given by a registered nurse, the FDA mandated that fospropofol, like propofol, must be used only in the presence of a trained anesthesia provider.
Introduction
The concept of moderate sedation and analgesia, introduced to replace the more arcane term conscious sedation, has been generally accepted in the anesthesia community as an appropriate target for sedation by non-anesthesiologists. Moderate sedation as defined by the American Society of Anesthesiologists (ASA) requires that the patient be arousable to verbal commands or light tactile stimulation. A patent airway, as well as stable cardiac and respiratory functions, are maintained throughout the period of sedation. The scope of monitored anesthesia care (MAC) is significantly wider, including the necessity of a preoperative evaluation, an anesthesiologist's personal participation or medical direction of the entire plan of care, and the ability to rescue a patient from unintended deep sedation or to intentionally provide deep sedation or general anesthesia if clinically warranted.
There is a common armamentarium of drugs shared between providers of moderate sedation and MAC, all given with the intent of maximizing anxiolysis and amnesia while maintaining a verbal patient. The ability of the patient to speak and understand is useful not only as a monitor of sedation depth and cardiorespiratory function but is also necessary to offer reassurance and communicate to the patient when active cooperation is required during the procedure (e.g. breath holding). 1 Propofol, a short-acting anesthetic agent that is rapidly titratable, is currently the premier agent chosen to achieve this purpose. Qualities such as a quick recovery time (even after a prolonged infusion) and the low incidence of nausea or emesis 2 have further augmented its popularity.
Propofol was introduced into clinical practice in 1986 by Astra Zeneca under the trade name Diprivan® (a shortened version of
Despite its widespread clinical use, propofol is not a drug that is free of unwanted side effects. Perhaps the most ubiquitous is pain at the injection site, 12 a phenomenon that is unreliably reduced by the addition of lidocaine to the propofol solution or by the injection of lidocaine into the vein prior to propofol. (The only technique shown to reliably reduce pain on injection in a majority (60%) of patients is to apply a tourniquet to the proximal arm and administer lidocaine 0.5 mg/kg 30-120 seconds prior to the propofol). 13 Propofol infusion syndrome, a rare but reported condition, includes severe metabolic acidosis, rhabdomyolysis, renal failure, and cardiac failure in association with a prolonged propofol infusion, critical illness, and the concurrent administration of catecholamines and steroids. 14 The lipid emulsion formula introduces another set of concerns including the need for absolute sterility when handling the drug, the relatively short window of usage (6 hours) when the vial is opened, and hypertriglyceridemia seen in patients receiving propofol infusion in the ICU setting.15–17 Finally, propofol has a remarkably narrow therapeutic window; even in trained hands, the dose curve bridging moderate sedation to general anesthesia may be unexpectedly steep. In susceptible patients, propofol is known to cause dose-dependent hemodynamic 18 and respiratory 19 depression and possibly loss of airway protective reflexes 20 in doses commonly used for mild to moderate sedation.
The stage was therefore set to develop a “kindler, gentler” propofol—one with less pronounced cardiorespiratory depression, preferably delivered in an aqueous form to eliminate the problems associated with the lipid emulsion. Investigators had proven that hydrophobic drugs could be made water-soluble by the addition of a large hydrophilic group, typically a phosphate monoester or a hemisuccinate, to create a prodrug. The hydrophilic addition was then enzymatically cleaved in vivo releasing the active drug. This approach has been used successfully with a variety of drug classes including antibiotics and steroids, 21 and more recently in the development of the anticonvulsant drug phosphenytoin. Aquavan® (Guilford Pharmaceuticals, Baltimore, MD), initially referred to as GPI 15715, was the first propofol prodrug adopted for clinical use. Chemically this water-soluble prodrug undergoes hydrolysis by alkaline phosphatase (predominantly at the endothelial cell surface) to release the active metabolite propofol, formaldehyde, and phosphate (Fig. 1). The liberated formaldehyde is rapidly converted to formate. Sedation and anesthesia are reliably produced among animals 22 as well as human 23 subjects.
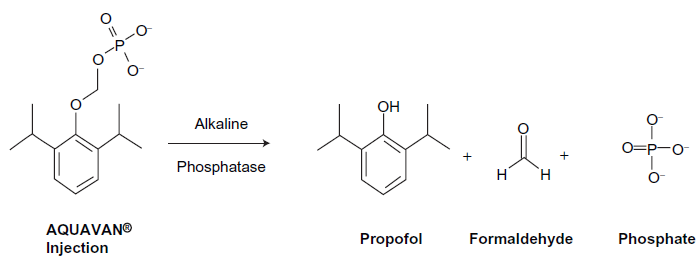
The conversion of fospropofol into its metabolites. 34
Pharmacokinetics
The pharmacokinetics of fospropofol have been extensively studied in both laboratory animals as well as humans, and details have been incorporated into a web based simulation comparing fospropofol to propofol. 24 In humans, a dual compartment model for the central distribution of the drugs was devised23,25,26 where the concentration of fospropofol in the central compartment was a function of the injected dose (D), and the concentration of propofol in the central compartment was a function of its conversion from fospropofol calculated in mass per time as F × kmet × CGPI × VCGPI, where F equals the fraction of the dose of fospropofol that is metabolized to propofol, kmet is the elimination rate constant of the prodrug, CGPI is its plasma concentration, and VCGPI is the volume of distribution of fospropofol in the central compartment. CGPI is measured directly, VCGPI and kmet are estimated, and F is calculated using the molecular weights of fospropofol and propofol (332 and 178, respectively) assuming a complete conversion. The data also implicated the presence of peripheral compartments for both drugs, and non-linear regression suggested the best fit for the data relied upon the presence of two peripheral compartments for each of the drugs, with transfer rate constants between the central and peripheral spaces designated as k12 and k21, k13 and k31, etc. (Fig. 2).
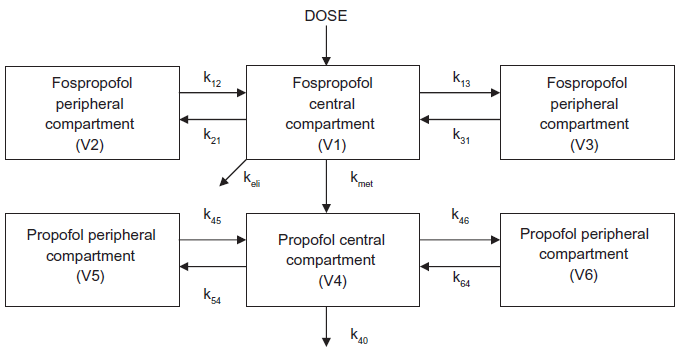
Some investigators suggest that fospropofol exists in a single peripheral compartment, thereby resulting in a five compartment model. Others 22 postulate a dual peripheral compartment for fospropofol, suggesting a six compartment model (shown).
The liberated propofol displayed typical lipophilic pharmacodynamics with large volumes of distribution. However, propofol derived from the parent prodrug showed significant differences in pharmacodynamic properties from propofol, namely a larger volume of distribution, lower peak plasma concentrations, and a shorter half-life due to a more rapid clearance. These differences were initially attributed to differences in sampling procedures and study design. It was later realized that an error in the assay 27 invalidated all of the quantitative pharmacokinetic data related to fospropofol. In all of the studies previously referenced samples of blood were collected in tubes containing a powdered form of sodium orthovanadate (SOV) to inhibit the alkaline phosphatase enzyme and therefore preclude the further conversion of fospropofol into propofol. Careful examination later revealed incomplete dissolution of the powder resulting in varying concentrations of SOV, thereby affecting plasma pH and in some instances causing hemolysis. Because these factors were neither known nor controlled at the time of the studies, all data derived relating to the propofol concentrations could therefore be inaccurate and is therefore now considered invalid. However the quantitative data relating to fospropofol itself is legitimate, as it was not affected by the assay. Repeated assays using liquid SOV, which would preclude this problem, have been suggested, but at the time of this writing they have yet to be published.
When fospropofol is converted to its active metabolite propofol, formate is released from the parent compound. In previous research high concentrations of formate have been shown to result in acidosis, ketonemia and acetonuria, respiratory compromise, and blindness. 28 In controlled studies it was demonstrated that no significant difference in intravenous formate levels existed between patients receiving fospropofol or Diprivan®. Furthermore, the level of intravenous formate was not found to vary with increasing doses of either of the induction agents. 25
Pharmacodynamics
Pharmacodynamic fospropofol studies in humans rely upon electroencephalographic evidence as well as clinical manifestations of the drug's effects. Dose escalation studies were performed on nine healthy male volunteers divided into 3 groups of 3 volunteers. 23 Each group received a fospropofol infusion over a 10 minute period, with Group 1 receiving a total dose of 290 mg each, Group 2 receiving 580 mg each, and Group 3 receiving 1160 mg each. The volunteers were tested for loss of consciousness (LOC), defined in this study by the absence of a response to a loud verbal command. If LOC was documented, the patient was further tested for a corneal reflex response, defined in this study as being a physical response to having a wisp of cotton rubbed across the cornea.
Among the members of Group 1 no LOC was documented. One patient reported an unpleasant sensation of tingling and burning in the anal and genital area lasting approximately 5 minutes which resolved without therapy. Among the 3 patients in Group 2, one patient experienced LOC 12 minutes after the initiation of the infusion and return to consciousness (ROC) was noted 22 minutes after the start of the infusion. Blood concentrations of propofol were obtained corresponding to LOC and ROC, but the aforementioned error in propofol analysis has invalidated the accuracy of these measurements. Amongst Group 3 all 3 patients displayed LOC 9 ± 3 minutes after the start of the infusion. ROC occurred 24 ± 2 minutes after fospropofol infusion was initiated. One patient again complained of a burning sensation in the anogenital region, spontaneously resolving after 2 minutes. Among the 4 patients who experienced LOC the administered dose of fospropofol was 870 ± 237 mg (mean ± SD). The corneal reflex was lost in only one patient, a member of the 1160 mg group.
All 9 subjects were evaluated using the Observer's Assessment of Alertness/Sedation Scale. 29 The scale was evaluated at 2, 5, 10, 20, 60, 120, and 240 minutes after the conclusion of the infusion (for patients who experienced no LOC) or after ROC. Patients were graded on a scale from 1 (deep sleep) to 5 (completely alert). Members of Group 1 achieved a score of 5 in 25 ± 5 minutes, Group 2 at 63 ± 49 minutes, and Group 3 at 112 ± 72 minutes.
The authors of the study also sought to measure the hemodynamic effects of fospropofol on the 9 volunteers. While one subject in Group 2 displayed an elevation in systolic blood pressure throughout the entire study period, the remaining subjects all showed a decrease in both systolic and diastolic pressures in the range of 20%-25%. Blood pressure values reached their nadir at 20 ± 8 minutes after the beginning of fospropofol infusion and returned to baseline approximately 60 minutes after the start of the infusion. All subjects showed an increase in heart rate, including one in Group 3 who had an elevation from 43 to 83 beats per minute. Heart rate reached its maximum value at 12 ± 8 min after the initiation of fospropofol infusion and returned to baseline at approximately 30 minutes after the start of the infusion.
In addition, respiratory and metabolic parameters were also measured. In all 9 subjects oxygen saturation was slightly reduced to a minimum value of 94.6% ± 1.6% reached 15 ± 3 minutes after the beginning of the fospropofol infusion (the duration of the period of decreased saturation was not specified in the original paper.) All 3 patients in Group 3 required insufflation of oxygen via a nasal cannula secondary to an oxygen saturation via pulse oximetry of less than 93%. Apnea was not observed in any of the subjects. An arterial blood sample drawn from each volunteer at the end of the infusion revealed a dose dependent rise in PaCO2 in the 3 ascending dosages. Body temperature remained constant in all subjects at 36.2 ± 0.4 C.
Fechner et al studied the efficacy of using a 2 hour infusion of fospropofol to induce a level of sedation that would theoretically be adequate for a minimally invasive procedure. 30 Their study group of 12 volunteers received a target controlled infusion (TCI) of the drug set to result in a propofol blood level of 1.8 μg/mL during the first hour. Again, their calculations rested upon data rendered inaccurate by the use of SOV and can only be evaluated qualitatively. Their goal during the first hour was to achieve a modified MOAA/S 31 (Table 1) score of 2 or 3 within 60 minutes. If any patient was outside of this range the dose would be adjusted either upward or downward during the second hour in an attempt to reach the target. Physiologic monitoring including BIS was performed throughout the study.
The Modified Observer's Assessment of Alertness/Sedation Scale.
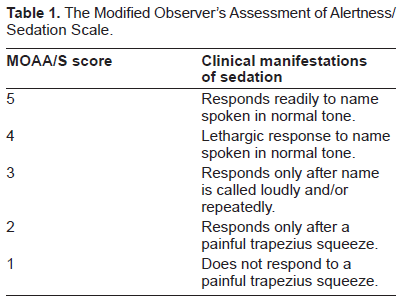
After 1 hour 9 of the 12 volunteers required an upward titration in their fospropofol infusions to achieve a satisfactory MOAA/S score. None required a downward titration due to oversedation. During the first hour the median MOAA/S score was 4 and the mean BIS was 72 ± 12. During the second hour the MOAA/S score dropped to a median of 3 with a decrease in the BIS to a mean value of 61 ± 11. Changes in systolic and diastolic blood pressure, heart rate, and oxygen saturation were consistent with previously published data. 11 of the 12 volunteers complained of genital and perianal paresthesias beginning 1 minute after the start of the infusion and resolving 2 minutes thereafter. 4 volunteers rated the sensation as mild and 7 reported it as moderate.
Although the pharmacokinetics of the study were flawed there are some salient points not apparent from the previously described fospropofol bolus studies. Among the nine patients who required an upward titration in their infusion, an increased level of sedation was reached at an average time of 3 minutes after the infusion rate was adjusted. This level of sedation was maintained during the remainder of the study. This suggests that fospropofol, like propofol, can be rapidly titrated in clinical practice to achieve a desired level of sedation. Recovery time after a 2 hour infusion was significantly higher for the fospropofol: after the 2 hour infusion the mean recovery time to a MOAA/S score of 5 was 18 minutes, approximately 10 minutes longer than for propofol. 32
Clinical Studies
Clinical studies with fospropofol have been conducted among patients receiving bronchoscopies or colonoscopies. In the former, the majority of the 500,000 flexible bronchoscopies performed annually in the United States 33 are performed under some type of intravenous sedation.34,35 While some authors have questioned the need for sedation during a flexible bronchoscopy, 36 the majority of clinicians use a combination of a benzodiazepine (to provide amnesia) and an opiod (to provide both analgesia and an antitussive effect). 37 One multicenter study has suggested that fospropofol may be a good alternative. 38 In this study, patients were randomized to receive either 2 mg/kg (non-therapeutic dose) or 6.5 mg/kg fospropofol prior to flexible bronchoscopy. All patients also received fentanyl 50 μg IV and topical lidocaine spray. Fospropofol was redosed every 4 minutes, up to three times, if the MOAA/S score was 5. The primary end point of the study was successful sedation (three consecutive MOAA/S scores < = 4) and successful treatment (the ability to complete the bronchoscopy without the use of additional sedatives or assisted ventilation.) Secondary end points included patient satisfaction with the procedure (willingness to undergo a repeat bronchoscopy with fospropofol), amnesia for the event, and time to recovery from sedation.
The higher dose group fared significantly better in achieving both primary end points. Among the 6.5 mg/kg group sedation success was 88.7% vs. 27.5% for the 2 mg/kg group (p < 0.001); treatment success also favored the higher dose group (91.3% vs. 41.2%, p < 0.001). The majority (56%) of the 6.5 mg/kg group required no supplemental doses of fospropofol, and only 8% required the addition of a benzodiazepine (compared to 7% and 58.8%, respectively, in the 2 mg/kg group.) The time until the patient was adequately sedated was 4 minutes in the high dose group vs. 18 minutes in the low dose group. Secondary endpoints also favored the higher dose: among the 6.5 mg/kg group, 94.6% would repeat the procedure with fospropofol and 83.3% had no recall of the event (vs. 78.2% and 55.4% respectively among the 2 mg/kg group, p < 0.001 for both.) Readiness for hospital discharge was slightly prolonged in the higher dose group (8.5 vs. 8.0 minutes) although the difference was statistically insignificant. (This should be contrasted with the 20-120 minute range for discharge readiness reported after the use of a benzodiazepine/opiod combination).39,40 Adverse events reported in both groups (pruritis, hypotension, and oxygen saturation below 92%) were rated by the patients and bronchoscopists as being mild to moderate and resolved either spontaneously or with minor intervention (e.g. increased oxygen flow, chin lift, fluid bolus.) The incidence of desaturation below 92% (15.4% in the high dose group, 12.6% in the low dose group) was equivalent to 41 or lower than prior published studies (24%-32%) 42 using a benzodiazepine/opiod combination.
The number of colonoscopies performed annually far eclipses the number of bronchoscopies due in part to the former's use as a screening tool as well as a diagnostic procedure. As with bronchoscopies, sedation has become the standard of care during colonoscopies 43 and has been shown to reduce the incidence of studies aborted prematurely due to patient intolerance.44,45 Gastroenterologists have become increasingly attracted to the use of propofol during colonoscopies. 46 Despite the package insert warning that propofol should be “administered only by persons trained in the administration of general anesthesia and not involved in the conduct of the surgical/diagnostic procedure” 5 gastroenterologists have been lobbying for this clause to be dropped, insisting that propofol can be safely administered by a registered nurse under the supervision of the physician performing the procedure.9,47–50 Both the American Society of Anesthesiologists and the American Association of Nurse Anesthetists have filed formal rebuttals arguing that the requested change is ill-advised. 51 The small but genuine incidence of adverse effects, coupled with the reluctance of payors to compensate for the anesthesia component of routine colonoscopies 52 has focused attention on fospropofol as being an all-purpose solution.
Clinical studies suggest that fospropofol may be as efficacious as propofol to provide sedation during routine outpatient colonoscopies. 53 One study showed both drugs provided significant reduced time to discharge (and associated economic savings). In the time to complete 1 colonoscopy using midazolam and meperidine (71.1 minutes), the clinicians were able to complete 1.76 colonoscopies using propofol and 1.91 using fospropofol, resulting in an increased profit margin of around $67 per colonoscopy in a hospital outpatient setting, and $57 per procedure in an ambulatory surgical center. 54 (These figures represent the profit for the gastroenterologist in the absence of an anesthesia provider. When such a provider is present and compensated, the average profits for the colonoscopy drop to $32 and $22, respectively). Savings attributed to rapid recovery were not analyzed from the perspectives of the patients (e.g. less need for childcare) and society (e.g. fewer days absent from work).
A common thread running throughout the GI literature is that propofol (due to its lack of analgesic properties) is insufficient alone to provide moderate sedation necessary for a successful colonoscopy. 55 The addition of a small dose of a benzodiazepine (which also has no analgesic properties) and/or an opiod (and possibly diphenhydramine as well) 56 has been shown to increase patient satisfaction, 57 reduce the dose of propofol by up to 50%, 58 and reduce the discharge time from the recovery area. 59 This practice has extended to include fospropofol, as evidenced by a study of patients receiving fospropofol for sedation during colonoscopies: all patients were premedicated with fentanyl 50 μg before initiation of sedation. Patients were then randomized to receive either fospropofol (2, 5, 6.5, or 8 mg/kg) or midazolam (0.02 mg/kg). 60 The goal was to maintain a MOAA/S score <=4; if necessary fospropofol was redosed every 4 minutes at ¼ of the original dose (for the fospropofol group) or midazolam was redosed every 2 minutes at 1 mg increments (for the benzodiazepine group.) Results were similar to the bronchoscopy study referenced earlier; patients in the 6.5 and 8 mg/kg groups had statistically significant greater success in sedation and treatment success when compared to their counterparts in the 2 and 5 mg/kg cohort (p < 0.001). Patients in the 8 mg/kg dose group were much more likely to enter a state of deep sedation (MOAA/S score of 0 or 1) vs. patients in the 6.5 mg/kg group (25% vs. 4%, respectively). Finally, patients in the 6.5 mg/kg group scored higher than any other group in measurements of patient satisfaction and willingness to repeat the procedure with the same method of sedation. No serious adverse effects were noted in any of the groups, and again the principle patient complaint was mild to moderate paresthesia.
In mid-December 2008 the FDA approved fospropofol for use in monitored anesthesia care settings. 61 Due to a series of corporate takeovers, fospropofol a.k.a. GPI 15715 a.k.a. Aquavan® will be marketed by the Eisai Corporation of North America under the trade name Lusedra®. Like propofol, the FDA has mandated that Lusedra® be used only by persons trained in the administration of general anesthesia, and that all patients should be continuously monitored by persons not involved in the conduct of the procedure. 62 The fact that fospropofol is not an induction agent has led some pulmonologists to feel safe circumventing the requirement for anesthesia personnel during its administration. 63 Other clinicians joined their chorus and lobbied for more liberal labeling. Despite the petitions, the DEA has classified Lusedra® as a Schedule IV controlled substance effective November 5, 2009. 64
Conclusions
Fospropofol may prove to be a useful tool for the anesthesia provider, offering many of the benefits of propofol while eschewing several of the concomitant side effects. The most prevalent side effect of fospropofol, genital and perianal itching, has not interfered with the widespread clinical adoption of other phosphorylated prodrugs (e.g. phosphenytoin) which share the same side effect profile. 65
The use of fospropofol to provide prolonged sedation in the intensive care unit remains controversial. While the Lusedra® package insert cautions against this practice, 66 reports exist about its successful utilization in mechanically ventilated patients in the intensive care unit setting. 67 While its use in this capacity will require further study, there is little doubt that fospropofol should prove to be a useful adjunct for anesthesia providers administering monitored anesthesia care in the operating room and other clinical sites.
Disclosure
Dr. Lubarsky has served on an advisory board for a company acquired by Eisai. Dr. Candiotti receives honorarium and research support from Eisai.