
Abstract
This case report describes a 5-year-old boy who presented to the emergency department with clinical symptoms and chest X-ray findings suggestive of pneumonia. Further history revealed multiple other infections, and workup for immunodeficiency revealed a deficiency of mannose-binding lectin (MBL), a pattern recognition receptor involved in activation of the complement system. Innate immunodeficiency may be more common than currently appreciated, with mutations of MBL affecting up to 50% of individuals in some populations. While pneumonia is a common presentation in the Pediatric Emergency Department, clinical presentations of children with defects of innate immunity can be unpredictable. Children may initially appear well with sudden deterioration. These cases pose particular challenges to physicians, and the level of suspicion for innate defects must remain high. It is crucial to identify patients with such impairments to better manage and prevent future complications.
Introduction
Pneumonia is a common presentation in the Pediatric Emergency Department. However, recent studies suggest that impairments of the innate immune system may account for this common presentation. It is crucial to identify patients with such impairments, to better manage and prevent future complications. In this paper, we present a case of a child who presented with pneumonia, in which the underlying defect was a mannose binding lectin (MBL) deficiency. This case exemplifies the need for a high level of suspicion, even in cases with mild initial clinical presentations.
Patient Presentation
A five-year-old boy presented to the emergency room (ER) with a 6-day history of fever, rhinorrhea, worsening cough, emesis, back and abdominal pain, decreased appetite, and headache. In the ER, he was found to have a temperature of 38.5 °C-39.5 °C, a respiratory rate of 60 breaths per minute, a heart rate of 140 beats per minute (bpm), blood pressure of 108/65, and an oxygen saturation of 96%-98% on 1/2 1pm of O2. He was tachypneic, with audible grunting, suprasternal retractions, a clear chest, mild tenderness at right upper and lower abdominal quadrants, but no guarding or rebound tenderness or hepatosplenomegaly, and no rashes. Laboratory values included a white blood cell count of 20 × 109/L, hemoglobin count of 122 g/L, platelet count of 462 × 09/L, absolute neutrophil count of 17 × 109/L, absolute lymphocyte count of 1.26 × 109/L, normal chemistry, elevated glucose of 9 mmol/L, urinalysis with glucose > 55 mmol/L and ketones. A chest X-ray (CXR) revealed right middle lobe pneumonia (Fig. 1). He was admitted to hospital and treated with intravenous ceftriaxone (75 mg/kg/day for 9 days), azithromycin (5 mg/kg/day po for 5 days) and vancomycin (20 mg/kg/day for 9 days, adjusted to a trough of 15–20).

Initial chest X-ray from the emergency department demonstrating a right middle lobe pneumonia.
Twenty-four hours later, due to persistent respiratory distress and increased chest pain, a repeat CXR was done and revealed extensive parenchymal involvement, mediastinal shift and a large effusion (Fig. 2). Ultrasounds of the chest and abdomen confirmed the pleural effusion, and revealed mild splenomegaly. In contrast to the degree of effusion, this child exhibited initially only moderate distress and did not appear toxic. A chest tube was inserted because of progressively worsening distress, and straw-colored pus was drained. The fluid was sent for cell count (white blood cell (WBC) 315, neutrophils 57%, platelet 7.5% red blood cell (RBC) 1210), protein (26), albumin (16), lactate dehydrogenase (LDH; 1308), lipase (17), amylase (32), gram stain (negative), culture (negative), streptococcal pneumonia polymerase chain reaction (PCR; negative), fungal culture (negative), mycobacterial culture (negative), viral culture (negative), and TB culture (negative). Blood drawn at the same time revealed a lipase of 16, amylase of 23, LDH of 168, and protein of 54. Other tests included a negative throat culture, a negative stool viral culture, negative blood culture, negative nasopharyngeal aspirate for respiratory viruses, antistreptolysin O titer (ASOT) of 134, antideoxyri-bonuclease-B (DNase B) titer < 50, and EBV titers suggestive of past infection. Nasopharyngeal aspirate was positive for mycoplasma pneumonia by PCR. There was a gradual improvement in his respiratory status during the following week and he was discharged after 9 days on amoxicillin-clavulanate (45 mg/kg BID for 7 days), followed by amoxicillin prophylaxis (250 mg po BID).
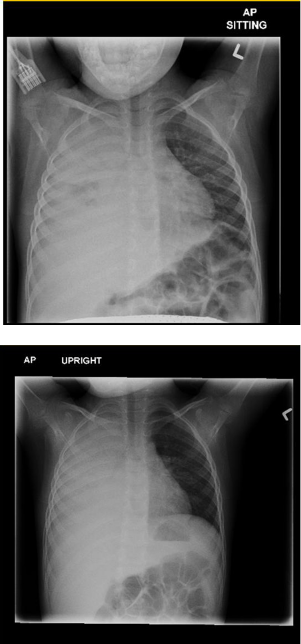
Chest X-ray 24 hours after admission revealing extensive parenchymal involvement, mediastinal shift and a large effusion.
Past medical history was significant for recurrent skin and respiratory infections, including a left elbow abscess requiring drainage and oral antibiotic treatment (at the age of 5 months), pneumococcal bacteremia presenting with fever and emesis (at the age of 7 months), a retropharyngeal abscess (at the age of 9 months), and pneumonia (at the age of 3 years). Developmental history was unremarkable, and current weight was 17 kilograms (> 15th percentile).
The parents are second cousins of Moroccan origin, and there is a healthy older brother.
Given the history of recurrent severe infections, he was initially evaluated at the age of 3 years by the immunology service at the Montreal Children's Hospital. His immune evaluation revealed a normal complete blood count (CBC), normal lymphocyte subsets, normal T cell proliferation to mitogens, normal serum IgG/A/M/E levels, and protective post-vaccination antibody titers to tetanus, diphtheria, hemophilus influenza, and streptococcus pneumonia. A dihydrorhodamine (DHR) test for phagocytic function was normal. Complement studies including CH50, AH50, C3, and C4 levels were normal, but there was absent activity through the mannose binding lectin (MBL) pathway for complement activation. With reproducible absence of functional MBL activity, DNA was sent for genetic analysis to identify known mutations associated with MBL deficiency. Results demonstrated a compound heterozygosity for two haplotypes in the MBL2 gene in this child. Both haplotypes identified, LYPB and LYQC, are associated with very low mannose binding protein levels. An additional polymorphism in the MBL promoter was also identified.
Given the history of significant infections associated with genetic and functional diagnosis of MBL deficiency, recommendations for management included antibiotic chemoprophylaxis, optimization of protection through routine vaccination, prompt investigation and treatment of infections, and regular immunology clinic visits to reduce the risk of long-term complications. At the time of his current admission to hospital the child was taking no regular medication, and despite repeated attempts of the immunology service to contact the family, he had been lost to follow-up. As a result, the family was not fully aware of the implications of his immunodeficiency and did not relay his underlying diagnosis to the ER staff.
Discussion
MBL leads to complement activation via the recognition of mannose residues present on the surface of bacteria. Deficiency of MBL has been associated with recurrent infections, failure to thrive, and diarrhea in children. 1
MBL, part of the innate defense against pathogens, is one of several pattern recognition receptors that identify repeating patterns of molecules such as carbohydrates found on microbial surfaces. MBL is an example of C-type lectin receptors that is synthesized in the liver, and circulates primarily in the serum although it has also been detected in middle ear fluid, synovial fluid of inflamed joints, amniotic fluid, and nasopharyngeal secretions. 2 Each MBL molecule is composed of 4 distinct regions: (1) a C terminal, carbohydrate recognition domain (CRD; lectin), (2) a collagen helix, (3) a hydrophobic neck region (α-helical coiled-coil domain), and (4) a cysteine-rich N-terminal region. Multiple chains interact to form dimers to hexamers in a bouquet-like structure. Although each CRD has a low affinity, the hexamer forms a structure with high avidity.1,2 MBL activates complement via MBL-associated serine proteases (MASPs). Following the binding of MBL to a microbial surface, MASP-2 is activated, cleaving C4 and C2, whose byproducts form the classical C3 convertase and thus initiates complement activation. MASP-1 may also cleave C3 directly, initiating the complement cascade 2 (Fig. 3).
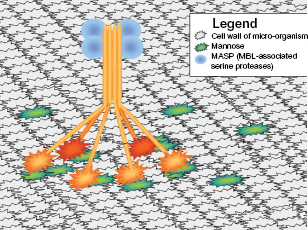
Mannose-Binding Lectin receptor, depicted as a hexamer, binding to mannose on the surface of a microbial cell wall. This pattern recognition receptor will activate the complement system via the MBL pathway, leading to the destruction of the micro-organism.
The wild-type MBL gene, designated MBL2 variant A, is located on chromosome 10 and, to date, three genetic mutations identified in exon 1 of this gene are known to result in low levels of circulating MBL. A mutation at codon 52 (arginine → cysteine) is denoted as variant D. Variant B (LYPB) results from a mutation in codon 54 (glycine → aspartic acid) and variant C (LYQC) is a glycine → glutamic acid mutation at codon 57. Other mutations in the promoter sequence have also been identified. 1 2 Heterozygous MBL mutations are relatively common—variant B is present in 22%-28% of the Eurasian population, variant C is present in 50%-60% of the sub-Saharan African population, and variant D is present in 14% of the European population. 1 These defects are thought to impair oligomerization of the MBL molecule and therefore result in functional MBL deficiency.
Low levels of MBL have been associated with susceptibility to specific infections. This is particularly the case in association with depressed immunity, such as is found in the neonatal period, neutropenia secondary to chemotherapy or infection, and chronic disease such as cystic fibrosis. 2 Further, MBL deficiency has been linked to septic shock and death. 1 The B variant allele is associated with symptomatic hepatitis B cirrhosis, spontaneous bacterial peritonitis, and, in patients with hepatitis C, lower success rates with interferon therapy. MBL may also play a role in human immunodeficiency virus (HIV) acquisition and spread, and in the development of acquired immune deficiency syndrome (AIDS). This may occur through the ability of MBL to opsonize HIV without neutralizing it, due to potential interference with virus trafficking and antigen presentation. In patients with mycoplasma infections, MBL deficiency was found to be more common than in a control group. 1 The typical patient with MBL deficiency is healthy, and its significance is appreciated when there is a co-existing immune defect. 1
Low MBL levels may have protective effects against intracellular pathogens such as Plasmodium falciparum, Cryptosporidium parvum, mycobacterial tuberculous meningitis, and Leishmania chagasi. 3 Intracellular parasites may use the C3 coating on their cells to enter host cells by means of the C3 receptor. Low levels of MBL can result in a reduced surface of C3 and thus impair the pathogen's ability to enter cells and complete their life cycle.1,4
It is possible that innate defects are much more common than is currently appreciated. Previous reports suggest that MBL activity in the immune response plays only a small role in protection against pathogens,6,7 yet our case suggests otherwise. We have shown here for the first time that MBL deficiency is associated with increased susceptibility to severe skin and respiratory infections. It is reasonable to speculate that the specific combination of mutations in MBL found in our patient, affecting both the coding gene sequence as well as its promoter, have resulted in substantial impairment in MBL function, and combined with the promoter sequence polymorphisms, account for the total absence of MBL and the severity of his clinical presentation. Indeed, homozygotes for the abnormal MBL alleles are reportedly predisposed to recurrent infections. 5 Hence, patients with recurrent pyogenic respiratory and cutaneous infections should be assessed for primary immunodeficiency, including defects in the MBL pathway, and where a functional MBL study suggests abnormal MBL activity, genetic testing may be required to further define the underlying immunodeficiency. In addition, this case exemplifies that the clinical progression of patients with innate immune defects are often unpredictable. Impairments in the innate system, as in our case, may be associated with mild initial signs of inflammation despite significant infectious load, due in part to the failure of innate immunity to signal through the inflammatory cascade. A high level of suspicion for innate defects should be maintained even in the absence of clinical symptoms suggesting severe illness. The patient described in this paper developed worsening respiratory distress while in hospital, necessitating chest tube insertion and drainage, despite the apparently lack of ancillary signs usually associated with an active inflammatory response. Thus, in patients with primary immunodeficiency associated with innate defects, treating physicians should be vigilant for the potential presentation of complications without antecedent clinical warning signs. Ongoing studies are needed to determine the true prevalence of such innate impairments.
Key Points
Heterozygous MBL mutations are relatively common in certain populations and homozygous cases might be at risk for severe infections.
MBL activates the complement pathway by way of mannose-binding lectin, a carbohydrate found on microbial but not human cell surfaces.
MBL deficiency can have unpredictable clinical consequences, and the absence of overt signs of inflammation does not rule out serious pathology.
A high index of suspicion should be maintained in patients who present with unusual or persistent infections.
Funding
This case presentation is part of the C-PRIMES initiative supported in part by an unrestricted research grant provided by CSL-Behring.
Competing Interests
CMcC is an advisory board member for CSL-Behring, Baxter and Talecris, has provided expert testimony for the US department of justice, and CMcC's institution has received grants from CIHR, CSL-Behring, Baxter and Griffols. Other authors disclose no competing interests.
Author Contributions
Analyzed the data: MH, MBS, CMC. Wrote the first draft of the manuscript: MH. Contributed to the writing of the manuscript: MH, MBS, CMC. Agree with manuscript results and conclusions: MH, MBS, CMC. Jointly developed the structure and arguments for the paper: MH, MBS, CMC. Made critical revisions and approved final version: MH, MBS, CMC. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Written consent was obtained from the patient(s) or their relative(s) to reproduce information or photographs appearing in this work.