
Abstract
Prostate cancer (PC) is one of the most common cancers and is a leading cause of death. Its initial growth is dependent on androgens; most patients show an initial response to hormonal therapy but will experience disease progression when PC becomes resistant to castration. In 2004, two key randomized controlled trials demonstrated a benefit for docetaxel-based regimens in the treatment of men with castration-resistant prostate cancer (CRPC). Cabazitaxel (XRP6258, TXD258, and RPR116258A), a tubulin-binding taxane drug as potent as docetaxel in cell lines, was the first treatment able to prolong survival for metastatic CRPC in the post-docetaxel setting. This review describes pharmacologic parameters of this agent followed by a review of clinical trials involving cabazitaxel. Other available treatments and the place of cabazitaxel in metastatic CRPC setting are discussed.
Introduction
Prostate cancer (PC) is one of the most common cancers in North America and Europe and is the second leading cause of male cancer-related death after lung cancer.1,2 The initial growth of prostate cancer is dependent on androgens. Since the discovery over 70 years ago that orchiectomy results in prostate cancer regression, androgen deprivation by the use of gonadotropin-releasing hormone (GnRH) agents (agonists or antagonists) or orchiectomy has been the foundation for the systemic treatment of metastatic prostate cancer. 3 Initial responses to chemical or surgical castration are quite favorable: this hormonal therapy leads to rapid biochemical responses, as assessed by declines in levels of the serum marker, prostate-specific antigen (PSA).4,5 However, most patients even if showing an initial response to hormonal therapy will experience disease progression after 12 to 24 months of treatment as evidenced by increasing PSA, radiologic progression, or progression of disease-related symptoms.6–8 “Androgen-insensitive” or “hormone-refractory” were terms previously used to describe prostate cancer progression despite medical or surgical castration. However, recent clinical observations and scientific work have shown that disease progression remains dependent on androgen receptor (AR) signaling and is sensitive to further hormonal manipulation; the term “castration resistant” is now preferred. This phase of the disease carries a much poorer prognosis.4,9,10 Prior to 2004, there was no treatment proven to improve survival for men with metastatic castration resistant prostate cancer (mCRPC). Prior to 2004, the main goal of chemotherapy by mitoxantrone combined with prednisone or hydrocortisone was to reduce pain and improve quality of life, but there was no benefit in terms of overall survival (OS).11,12 In 2004, two phase 3 trials, TAX 327 and SWOG (Southwest Oncology Group) 9916, showed a benefit for docetaxel-based regimens in the treatment of men with CRPC.13,14 Untill 2010, no treatment has demonstrated survival improvement for patients whose disease progresses after docetaxel treatment. Mitoxantrone was often administered because of its favorable effects on quality-of-life outcomes.13,14 Cabazitaxel (XRP6258; TXD258; RPR116258A), a tubulin-binding taxane drug as potent as docetaxel in cell lines, 15 was the first treatment able to prolong survival for metastatic castration resistant prostate cancer in the post-docetaxel setting. 16 This review describes pharmacologic parameters of this agent, then clinical trials involving cabazitaxel (CAB) and other available treatments in mCRPC are discussed.
Pharmacology
Mechanism of Action
Taxanes have represented a new major class of chemotherapy agents over the last two decades as shown by their extensive use as single agents and in multiagent regimens to treat various solid malignancies.17,18 However, their high substrate affinity for the multi-drug resistance (MDR) proteins represents one potential limitation. Taxanescan confer both constitutive and acquired resistance.19,20 The new taxane cabazitaxel was selected for clinical development due to its low affinity for the ATP-dependent drug efflux pump, P-glycoprotein 1 (P-gp). In addition, this compound has greater penetration of the blood-brain barrier compared with docetaxel and paclitaxel. 21 Cabazitaxel (formula C45H57NO14) is partially synthesized as a single diastereoisomer from 10-deacetylbaccatin III, the major natural taxoid derived from the needles of various Taxus species. It promotes tubulin assembly and microtubules stabilization against cold-induced depolymerization in vitro as potently as docetaxel.21,22
Docetaxel cytotoxicity was compared with cabazitaxel in several murine and human cell lines. 21 Cabazitaxel showed potent antitumor activity comparable with docetaxel in docetaxel-sensitive cell lines, with 50% tumor inhibition at concentrations ranging from 0.003 to 0.029 μmol/L. Moreover, cabazitaxel was more potent than docetaxel in various cancer cell lines with acquired resistance to docetaxel due to P-gp overexpression, including P388/TXT, P388/VCR, Calc18/TXT, P388/DOX, HL60/TAX, and KBV1. 21 Resistance factor ratios ranged from 1.8 to 10 for cabazitaxel, whereas comparable values were 4.8 to 50.7 for docetaxel. Furthermore, cabazitaxel showed increased cytotoxicity compared with docetaxel in a human colon adenocarcinoma cell line (CaCo-2) that exhibits primary resistance to the taxanes due to MDR. 23 In mice bearing implanted human xenografts, broad spectrum of antitumor activity has been shown for cabazitaxel with various cell lines: HCT116 colon, A549 lung, MIA PaCa-2 pancreatic, SR475 squamous cell, and Du-145 prostate cancers.21,22
Another preclinical study suggested a nonlinear accumulation of cabazitaxel in the brain of rodent. 24 It seems to occur by saturation of the P-gp at the rodent blood-brain barrier. This saturation could have several advantages, such as overcoming a P-gp–mediated efflux and could be usefull in case of brain metastases. However, the nonlinear pharmacokinetics could increase the risk of toxicity.
Pharmacokinetic (PK) and Metabolism Profile
The pharmacokinetics of cabazitaxel are linear in the studied dose range of 10 to 30 mg/m2 given as 1-hour infusions and are consistent with a three-compartment PK model. 25 Cabazitaxel PK profile was similar to that of docetaxel and characterized by doses proportionality in the dosing range of the Mita phase 1 study and triphasic elimination in plasma. 22 After cumulative treatment in patients in whom plasma sampling was performed during multiple courses, no evidence of major changes in the PK behavior of cabazitaxel was found. This suggests the absence of autoinduction or drug accumulation in plasma. At steady state, cabazitaxel distribution volume seems larger than that of docetaxel (mean Vss values, 2034 ± 1,495 versus 83.2 L/m2), and its terminal half-life is longer (mean t1/2 λ 3, 77.3 ± 45.5 versus 11.2 hours, respectively.22,26 The interpatient variability in the phase 1 was moderate and estimated at 40.7% of AUC(0–48 h). 22 Results of a population PK model (developed and validated with data from 170 patients treated with cabazitaxel included in five studies) indicated that interindividual variability of cabazitaxel clearance was significantly related to body surface area and tumor type. 27
Cabazitaxel is mainly metabolized by cytochrome P450 (CYP) 3A4 and CYP3A5 (the contribution of CYP3A estimated to be in the range of 80%–90%) and to a lesser extent by CYP2C8. The metabolism of cabazitaxel may be modified by the concomitant administration of drugs that are known inhibitors (eg, ketoconazole) or inducers (eg, carbamazepine, phenobarbital, rifampicin, and phenytoin) of CYP3A. Moreover, cabazitaxel administration with compounds known to be primarily metabolized through CYP3A may increase the exposure of these medicinal products. Of note, CYP3A4 and CYP3A5 are subject to genetic polymorphism. 28 In a population pharmacokinetic analysis in 70 patients aged ≥ 65 years (57 were aged 65–75 years and 13 were aged > 75 years), no age effect on the PKs of cabazitaxel was observe. Cabazitaxel is contraindicated in patients with hepatic impairment (bilirubin ≥ 1 × the upper limit of normal [ULN] or aspartate aminotransferase and/or alanine aminotransferase ≥ 1.5 × ULN). Mild to moderate renal impairment did not have meaningful effects on the pharmacokinetics of cabazitaxel. No data are available for patients with severe renal impairment (creatinine clearance < 30 mL/minute) or end-stage renal disease; therefore, these patients should be treated with caution and monitored carefully during treatment. 28 Cabazitaxel was largely excreted in the feces (63% to 77% of the dose), whereas the urinary route contributed markedly less (3% to 4% of the dose) over 2 weeks. 27 A rapid and sensitive liquid chromatography/tandem mass spectrometry (LC–MS/MS) method has been developed and validated for the quantitative determination of cabazitaxel. This method will prove to be a valuable tool for pharmacokinetic (interaction) studies with cabazitaxel. 25
Clinical Studies
Preliminary Studies
Mita et al conducted the first phase 1 study involving cabazitaxel. 22 The objectives of this phase 1 study were to characterize the toxicities of cabazitaxel, determine the maximum tolerated dose (MTD) and the recommended dose for phase 2 studies, characterize the PK profile of the compound, and document preliminary evidence of antitumor activity. Cabazitaxel was administered as 1-hour intravenous (IV) infusion every 3 weeks. Patients with histologically documented advanced solid malignancies refractory to conventional treatment could be candidates for this trial. Patients should have received less than 2 prior chemotherapy regimens for metastatic disease and/or radiotherapy affecting < 25% of their hematopoietic reserve to be eligible. The starting dose was 10 mg/m2. This dose corresponds to approximately one tenth of the severe toxic dose (STD10) in mice and to the single highest nonseverely toxic dose in dogs. Then subsequent dose levels could increase to 15, 20, and 25 mg/m2. Between 1999 and 2001, 25 patients were treated with 102 courses of cabazitaxel across four dose levels. All 25 patients (100%) were evaluable for safety and 24 patients (96%) were evaluable for efficacy. At 20 mg/m2, dose-limiting toxicity (DLT) was not observed in the initial three patients enrolled. However, three of seven subjects experienced DLT, including febrile neutropenia in one minimally pre-treated (MP) patient and protracted (>5 days) grade 4 neutropenia in two heavily pretreated (HP) patients at 25 mg/m2. Nonhematologic toxicities were generally mild to moderate in severity. They included nausea, vomiting, diarrhea, neurotoxicity, and fatigue. 22 Concerning its activity, the authors described partial responses (PRs) in two patients with metastatic prostate carcinoma, one unconfirmed PR, and two minor responses. At the 25 mg/m2 dose level, the rate of DLT exceeded the predefined limits of tolerability. No DLT was observed in six additional MP and HP patients treated at the previous dose level, 20 mg/m2. This last dose level was considered the recommended phase 2 dose for both MP and HP patients. 22
There is no published phase 2 study in a PC setting. However, two phase 2 studies in patients with metastatic breast cancer were described here to specify the optimal dose of cabazitaxel and its safety profile.
The first multicenter phase 2 study assessed its activity in the treatment of taxane-resistant metastatic breast cancer. 29 Cabazitaxel was administered as a 1-hour IV infusion every 3 weeks at 20 mg/m2 (then, in the absence of severe toxicity, at 25 mg/m2 from cycle 2). The primary end point was the objective response rate (ORR) assessed according to response evaluation criteria in solid tumors (RECIST) guidelines. Seventy-one patients were included with a median relative dose intensity of 0.98. Objective antitumor activity included an ORR of 14% (two complete, eight PR) with 18 patients (25%) who had stable disease of >3 months duration. The median time to progression was 2.7 months, and the median overall survival (OS) was 12.3 months with a median follow-up of 20.0 months. Neutropenia was the most common grade 3/4 hematological adverse event (AE) occurring in 73% of patients and 43% of evaluable cycles. Treatment-related febrile neutropenia or neutropenic infections were observed in 3% and 4% of patients and in <1% of cycles, respectively. Grade 3/4 anemia and thrombocytopenia were rare. Dose escalation up to 25 mg/m2 from cycle 2, in selected patients, on the basis of their good tolerance in cycle 1, was feasible only in 20 patients (28%) with no evident increase in the overall incidence of subsequent AEs in this group. Treatment emergent grades 3 to 5 nonhematological AEs probably or possibly related to study treatment were also rare, with the most common including hypersensitivity (4%), fatigue (3%), and hemorrhagic cystitis (3%). No severe AE occurred for nausea, vomiting, neuropathy sensory, myalgia and fluid retention. The most frequent nonhematological AEs at any grade, occurring in >15% of patients, were fatigue (35%), nausea (32%), diarrhea (30%), vomiting (18%), myalgia (17%), neuropathy sensory (17%), and anorexia (15%). Two deaths were reported, one related to the study drug and one to an unknown cause. Cabazitaxel was active and well tolerated in this group of metastatic breast cancer patients with taxane-resistant disease. 29
The second study was conducted by Vilanueva et al. 30 The objectives of this phase 1/2 study were to assess the MTD, safety profile, pharmacokinetics, and activity of cabazitaxel plus capecitabine in patients with metastatic breast cancer who had been previously treated with taxanes and anthracyclines. This was a two-part study: in part one, a 3 + 3 dose–escalation scheme was used to assess the MTD of intravenous cabazitaxel (day 1) given with oral capecitabine twice daily (days 1–14) every 3 weeks. In part two, the ORR of the combination at the MTD was evaluated. Thirty-three patients were included and treated with 15 patients in part one and 18 in part two. Cabazitaxel 20 mg/m2 combined with capecitabine 1000 mg/m2 was the MTD. Pharmacokinetic analysis did not show apparent drug–drug interaction. An interpatient variability in all PK parameters for cabazitaxel was observed (52%–69%). Including all patients, the main grade 3 to 4 toxicities were: neutropenia (n = 21), hand–foot syndrome (n = 5), asthenia (n = 5), neutropenic infection (n = 1), and neutropenic colitis (n = 1). One patient had febrile neutropenia. Antitumor activity was observed at all dose-levels: 2 complete responses, 5 PRs, and 20 disease stabilisations (7 unconfirmed PR). At the MTD, 21 patients were evaluable for efficacy with an ORR of 23.8% (95% confidence interval [CI], 8.2%–47.2%). The median response duration was 3.1 months (95% CI, 2.1–8.4 months) and median time to progression was 4.9 months. 30
Phase 3 Randomized Study in mCRPC Patients: Tropic Trial
Based on the results from the phases 1 and 2 mentionned above, one could recommend an optimal dose of cabazitaxel administered as a 1-hour IV infusion every 3 weeks at 20 mg/m2 for further phase 3 clinical trials. However, it is not the schedule that was chosen in the international randomized open-label phase 3 TROPIC trial. 16 Patients were centrally randomly assigned to receive cabazitaxel 25 mg/m2 with premedication (antihistamine, corticosteroid) intravenously over 1 hour or mitoxantrone 12 mg/m2 intravenously over 15 to 30 min on day 1 of each 21-day cycle and were stratified for disease measurability (measurable vs. nonmeasurable) and ECOG performance status (0–1 vs. 2). The primary endpoint was overall survival. Patients had pathologically proven prostate cancer with documented disease progression during or after completion of docetaxel treatment. An amendment was made to the trial protocol after 59 patients had been enrolled to exclude patients previously receiving a cumulative docetaxel dose lower than 225 mg/m. Eligible patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2. Patients with nonmeasurable disease were required to have rising serum prostate-specific antigen (PSA) concentrations or the appearance of at least one new demonstrable radiographic lesion. All patients received oral prednisone 10 mg daily.
Between 2007 and 2008, 755 patients were randomly assigned to treatment groups (378 cabazitaxel and 377 mitoxantrone). Patients characteristics are summarized in Table 1. Roughly 50% of patients had measurable soft tissue disease and 25% had visceral (poor prognosis) disease. The median dose of docetaxel received before the study was 576.6 mg/m. (interquartile range [IQR] 408.4–761.2) in the cabazitaxel group and 529.2 mg/m. (IQR 380.9–787.2) in the mitoxantrone group. About 70% of patients had progressive disease either during or within 3 months of completing docetaxel treatment, including about 30% of patients who had disease progression during docetaxel treatment. The median follow-up for both treatment groups combined was 12.8 months. The Kaplan-Meier analysis (Fig. 1) showed an OS benefit in favor of cabazitaxel with a median OS of 15.1 months (95% CI, 14.1–16.3) versus 12.7 months (11.6–13.7). This result corresponds to a 30% reduction in relative risk of death (hazard ratio [HR], 0.70; 95% CI, 0.59–0.83;
Patients’ characteristics in the phase 3 clinical trials implicating cabazitaxel, abiraterone and MDV3100 in mCRPC patients previously treated by docetaxel.


Kaplan-Meier estimates of the probability of overall survival (OS) and progression free survival (PFS) in the phase 3 TROPIC trial. Figure adapted from ref 16 (de Bono et al).
Activity and safety profile of the phase 3 clinical trials implicating cabazitaxel, abiraterone and MDV3100 in mCRPC patients previously treated by docetaxel.
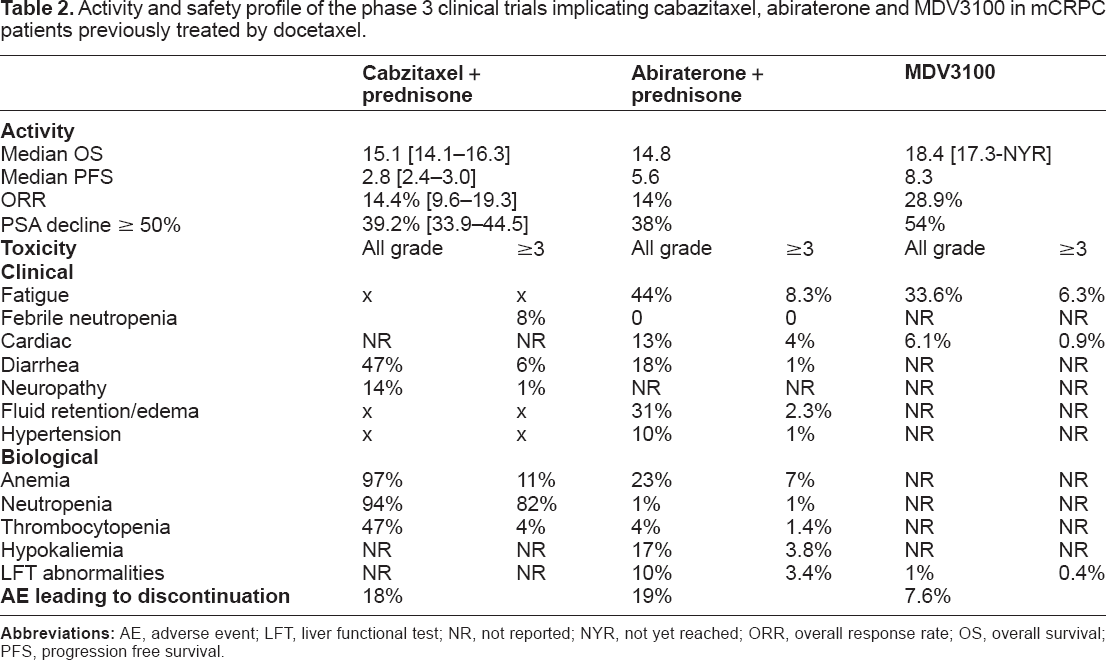
The most common toxic effects of cabazitaxel were haematological; the most frequent haematological grade 3 or higher AEs were neutropenia, leukopenia, and anaemia (Table 2). The most common nonhaematological grade 3 or higher AE was diarrhoea. Grade 3 peripheral neuropathy was infrequent (reported in only three [1%] patients in each group). Overall, all grades peripheral neuropathy and peripheral edema were reported during the study in 52 (14%) and 34 (9%) patients in the cabazitaxel group and 12 (3%) and 34 (9%) in the mitoxantrone group, respectively. 18 (5%) patients treated with cabazitaxel and nine (2%) treated with mitoxantrone died within 30 days of the last infusion. The primary reason for treatment discontinuation in both groups was disease progression. Dose reductions were reported for 45 (12%) patients in the cabazitaxel group and 15 (4%) mitoxantrone-treated patients, and treatment delays occurred in 104 (28%) and 56 (15%) patients, respectively. Overall, 5% of mitoxantrone treatment courses were dose reduced compared with 10% of cabazitaxel treatment courses. 16
The findings of TROPIC established cabazitaxel as the first agent to prolong survival in the post-docetaxel space, with a 30% reduction in death over mitoxantrone. 16 The rate of febrile neutropenia in the cabazitaxel group was 8%, suggesting that cabazitaxel treatment in this noncurative setting requires careful monitoring and management of emerging symptoms. Dose modifications (delay or reductions) as well as prophylactic treatment with granulocyte colonystimulating factor in high-risk selected patients are potential risk-mitigation strategies that could be considered to manage these toxic effects. On the basis of these data, cabazitaxel was granted fast track designation by the United States Food and Drug Administration (FDA) in November 2009. In March 2011, the European Medicines Agency (EMA) 27 adopted a positive opinion to grant a marketing authorization in the Europeen Union for Cabazitaxel.
Other Therapies in mCRPC Setting
Since 2004, the use of a docetaxel-prednisone regimen as first-line chemotherapy is considered a standard of care for men with mCRPC. In the past 2 years, the landscape has changed rapidly. Results from phase 3 trials with new coumponds have become available, resulting in the introduction of various new approaches predocetaxel and postdocetaxel.
Hormonal Therapy
Recent progress has been achieved in the past decade on the hormonal side of PC. Studies suggest that recurrent PC despite castrate serum testosterone levels is not truly androgen-independent. It has been found in the prostate of men with CRPC that androgen levels still remain nearly equivalent to those in noncastrate patients. 31 These androgens seem to be produced directly in prostate cancer cells due to an upregulation of enzymatic pathways.32,33 Several other mechanisms involved in the malignant activation of AR in prostate cancer by castrate levels of androgen include mutations of the AR that can affect its ligand promiscuity, increased AR expression, and molecular cross-talk with other signaling pathways and co-regulators that lie downstream.5,34,35,36 The clinical translation of these concepts was confirmed by the results of two randomized phase 3 trials with new hormonal therapies: the androgen biosynthesis inhibitor abiraterone acetate and a novel AR antagonist MDV3100. In both of these trials, improved overall survival was demonstrated in a population of patients with disease progression following first-line docetaxel chemotherapy.37,38 Patient characteristics, activity, and safety parameters are detailled in Tables 1 and 2.
Immunotherapy
The first immunotherapy for the treatment of prostate cancer that had been FDA approved was sipuleucel-T, an autologous activated dendritic cell therapy, given as 3 consecutive infusions every 2 weeks. Sipuleucel-T is an autologous active cellular immunotherapy, consisting of patients’ autologous peripheral blood mononuclear cells (PBMCs) stimulated ex vivo with PAP-GM-CSF, a recombinant protein consisting of the target antigen prostatic acid phosphatase (PAP) fused to granulocyte macrophage colony-stimulating factor (GM-CSF). After reinfusion, this strategy aims at stimulating an effective immune response against human PAP, an antigen highly expressed in prostate cancer tissue. Three randomized, double-blind, controlled, multicenter phase 3 studies (D9901, D9902A and D9902B) enrolled a total of 737 patients.39–41 The IMPACT (Immunotherapy Prostate Adenocarcinoma Treatment) trial, in which 512 chemotherapy naive patients with CRPC were randomly assigned in a 2:1 ratio to either sipuleucel-T or placebo, reported an overall survival benefit of 4.1 months (25.8 vs. 21.7 months; HR, 0.78; 95% CI, 0.61–0.98;
Other novel forms of immunotherapy being tested in patients with CRPC include the use of anti-CTLA4 (Cytotoxic T-lymphocyte–associated antigen 4) blockade with ipilimumab and immunization with PROSTVAC-VF, a poxviral-based PSA-targeted vaccine.
42
A recently published randomized, controlled, double-blind, phase 2 study of PROSTVAC-VF including 125 patients with chemotherapy-naive minimally symptomatic metastatic CRPC and Gleason score of <7 showed promising results.
42
There was no improvement in progression-free survival (PFS), the primary endpoint of the study, but patients receiving PROSTVAC-VF experienced a median survival benefit of 8.5 months (25.1 vs. 16.6 months for controls; HR, 0.56, 95% CI, 0.37–0.85,
Bone Targeted Therapy
The bone is an important target in advanced metastatic prostate cancer since most patients will develop bone metastases during the course of their disease, and most disease-related symptoms are directly related to bone metastases. Bone metastases are the main cause of significant morbidity and poor quality of life, and may hasten death; it represents an important therapeutic target in such disease. Bisphosphonates such as zoledronic acid have demonstrated utility at preventing skeletal complications in patients with CRPC with bone metastases.
52
Zoledronic acid (4 mg via a 15 min infusion every 3 weeks for 15 months) reduced the incidence of skeletal-related events (SREs) in men with hormone-refractory metastatic prostate cancer. The receptor activator of nuclear factor-κB ligand (RANKL) inhibitor, denosumab, has been developed for the treatment of bone metastases. RANKL is involved in the regulation of bone metabolism and is overexpressed in osteoblasts. A phase 3 randomized noninferiority trial was performed in 1904 men with bone metastases from CRPC and no previous exposure to intravenous bisphosphonate. It compared denosumab and zoledronic acid with end point time to first SRE.
53
Denosumab was better than zoledronic acid for prevention of SREs median time to first on-study SRE was 20.7 months (95% CI, 18.8–24.9) with denosumab compared with 17.1 months (95% CI, 15.0–19.4) with zoledronic acid (HR, 0.82; 95% CI, 0.71–0.95;
Conclusion
In the last 2 years, five new treatments for CRPC (sipuleucel-T, cabazitaxel, abiraterone acetate, alpharadin, and now MDV3100) have shown a survival benefit in randomized trials (Fig. 2). This has allowed approval by the United States FDA and the European Medicines Evaluation Agency (EMEA) for some of them. In April 2010, the FDA approved an autologous cellular vaccine, sipuleucel-T (Provenge) for the treatment of metastatic CRPC due to demonstrated OS benefit. Currently, three novel molecucles have been approved by the FDA and EMEA: cabazitaxel (Jevtana) and abiraterone (Zytiga) were approved for the treatment of patients with metastatic CRPC postdocetaxel, and denosumab (XGEVA) was approved for the supportive management of bone disease. Patients are now living with advanced prostate cancer for longer with improved quality of life and better palliation of symptoms. Median OS at the initiation of chemotherapy has ranged from 12.3 months at the end of 1990s to 29 months with squential treatment with docetaxel and cabazitaxel.12,57 The optimal dose of cabazitaxel is still to be further explored: two phase 3 studies are recruiting patients. The first one, PROSELICA trial (NCT01308580), aims at demonstrating the noninferiority in terms of overall survival (OS) of cabazitaxel 20 mg/m2 (arm A) versus cabazitaxel 25 mg/m2 (arm B) in combination with prednisone in patients with metastatic castration-resistant prostate cancer (MCRPC) previously treated with a docetaxel-containing regimen. The place of cabazitaxel versus docetaxel will be precised with the second trial, FIRSTANA (NCT01308567). This trial aims at demonstrating the superiority of cabazitaxel plus prednisone at 25 mg/m2 (arm A) or 20 mg/m2 (arm B) versus docetaxel plus prednisone (arm C) in term of overall survival (OS) in patients with metastatic castration-resistant prostate cancer (mCRPC) and not previously treated with chemotherapy.
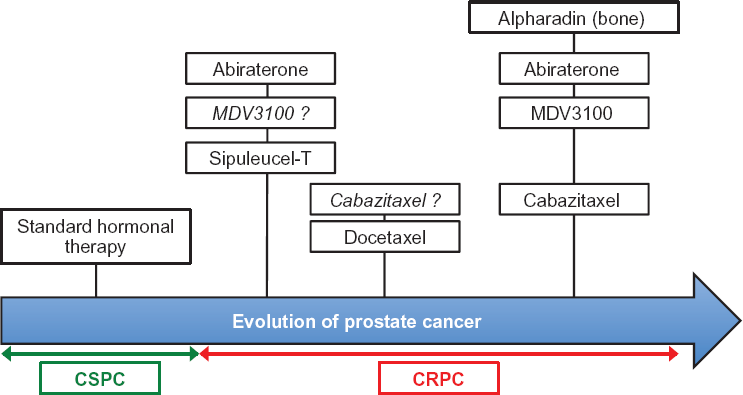
Drugs having shown a benefit according to their primary objective in phase 3 trials in patients with CRPC. Abiraterone in predocetaxel setting had 2 primaries and has demonstrated a significant radiological progression free survival benefit and a strong trend in overall survival benefit. Drugs in italic = phase 3 on-going.
The place of chemotherapy remains to be defined: recent results presented at the annual meeting of American Society of Clinical Oncology (ASCO) 2012 demonstrated a significant radiological PFS benefit and a strong trend in overall survival benefit of abiraterone plus prednisone over placebo plus predisone for patients with asymptomatic or mildly symptomatic mCRPC. 58 Other phase 3 studies exploring the impact of docetaxel based chemotherapy when used in earlier stage have shown positive biological PSA responses at the ASCO 2011 meeting.59–61 Clinical events are strongly awaited. Many agents (bevacizumab, aflibercept, lenalidomide, zibotentan, atrasentan, and calcitriol) failed to show a benefit when added to docetaxel base-chemotherapy. Results of phase 3 trials exploring the association of docetaxel with dasatinib or OGX-011 will be available probably in 2013. The place of cabazitaxel in the mangement of CRPC will depend on sequential strategies that will involve all these new molecules. Results of the FIRSTANA trial, if positive, could make cabazitaxel more usefull in earlier stage. With the range of newer treatment options becoming available, it is clear there will be a need to more carefully define the most appropriate sequence of treatment for individual patients with CRPC.
Author Contributions
Analysed the data: FC, TNG, ED, CV, EC, SK, PM, FK, PX, ATV. Wrote the first draft of the manuscript: FC, TNG, ED, ATV. Contributed to the writing of the manuscript: CV, EC, SK, PM, FK, XP. Agree with manuscript results and conclusions: FC, TNG, ED, CV, EC, SK, PM, FK, PX, ATV. Jointly developed the structure and arguments for the paper: FC, TNG, ED, ATV. Made critical revisions and approved final version: FC, TNG, ED, CV, EC, SK, PM, FK, PX, ATV. All authors reviewed and approved of the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
FK is a board member for Sanofi-Aventis, Janssen and Ferring, and has received payments for lectures from Steba Biotech, Ipsen, Takeda, Sanofi-Aventis, and is a trial investigator for Steba Biotech and Ferring. XP has received consulting fees/honorarium from Sanofi, and is also a board member. A-TV is a board member for Sanofi, Novartis, Ferring and Astellas, and has received payment for lectures from Roche, Ipsen, and Takeda. Other authors disclose no competing interests.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Provenance: the authors were invited to submit this paper.