
Abstract
Chemotherapy using the taxanes, docetaxel and cabazitaxel, remains an important therapeutic option in metastatic castration-resistant prostate cancer (CRPC). However, despite the survival benefits afforded by these agents, the survival increments are modest and resistance occurs universally. Efforts to overcome resistance to docetaxel by combining with biologic agents have heretofore been unsuccessful. Indeed, resistance to these taxanes is also associated with cross-resistance to the antiandrogen drugs, abiraterone and enzalutamide. Here, we discuss the various mechanisms of resistance to chemotherapy in metastatic CRPC and the potential role of emerging regimens and agents in varying clinical phases of development.
Introduction
Prostate cancer is estimated to occur in 180,890 men in the USA in 2016 alone. 1 Prostate cancer has been recognized since 1942 as being responsive to androgen deprivation therapy (ADT), but the response may be transient and the majority of patients progress to castration-resistant prostate cancer (CRPC) by developing resistance to ADT by various mechanisms. 2 Approximately 20% of the patients may have de novo resistance to ADT. 3 Chemotherapy has played a pivotal role in the setting of metastatic CRPC (mCRPC) since 2004 when docetaxel-based chemotherapy first showed modest improvement in survival compared with mitoxantrone-based therapy as first-line chemotherapy.3–5 Older agents, including estra-mustine, platinums, cyclophosphamide, and 5–fluorouracil, exhibit marginal-to-modest activity but have not been pursued in randomized trials.6–9 Mitoxantrone in combination with low-dose corticosteroids confers palliative benefits without overall survival (OS) benefit.3,10–13
After a hiatus since the approval of docetaxel, multiple new orally administered antiandrogen drugs, such as abiraterone acetate and enzalutamide, have extended survival in the pre- and postdocetaxel settings.12,14–16 Additionally, an immunotherapeutic agent (sipuleucel-T) and a radiopharmaceutical (radium 223) have also extended survival modestly in minimally symptomatic or symptomatic bony disease settings, respectively.17,18 Moreover, a second and more potent taxane, cabazitaxel, has extended survival in postdocetaxel patients. 19 The oral antiandrogen drugs have been rapidly adopted in the clinic due to the favorable therapeutic indexes and convenience, and chemotherapy has been generally relegated to later-line settings.
Cross-resistance between antiandrogen drugs and taxanes, as well as resistance to chemotherapy, is emerging as an important barrier to be overcome. Understanding the mecha-nisms of chemoresistance is of utmost importance as it can help in developing newer therapeutic agents and potentially synergistic combinations with better efficacy and improved survival. 2 In addition, identifying patients who are potentially not likely to benefit from chemotherapy can prevent substantial morbidity in those patients. One challenging issue is that resistance is identified on the basis of disease progression, which is a soft end point in mCRPC, due to the frequent presence of nonmeasurable bone metastases. Indeed, the Prostate Cancer Working Group guidelines recommend 3 months of therapy before objective assessment, due to the occurrence of early prostate-specific antigen (PSA) and bone scan flares. 20 Additionally, switching therapy based solely on PSA changes is not recommended. This review describes the current role of chemotherapy for treating mCRPC and mechanisms of resistance to chemotherapy.
Docetaxel
Mechanism of action
Docetaxel is an antimitotic agent historically recognized to inhibit microtubule disassembly and has more recently been demonstrated to downregulate androgen receptor (AR) transcriptional activity. Docetaxel can inhibit the translocation of the AR to the nucleus in response to both androgens and ligand-dependent signaling path-ways.21,22 Docetaxel also inhibits AR gene expression by acting on the gene promoter. It increases the levels of Forkhead box O1 (FOXO1), a strong transcriptional repressor of AR. 23 The downregulation of AR activity, in addition to its historically recognized antimitotic effects, can explain the efficacy of microtubule inhibitors in prostate cancer. 21 In addition, docetaxel also has anti-B-Cell lymphoma (BCL)-2 and anti-BCLX properties, thereby promoting apoptosis.21,24 Two Phase III clinical trials established docetaxel-based chemotherapy as the standard first-line chemotherapy based on an ~3-month increment in median OS compared to the mitoxantrone arm, which led to approval of the drug by the US Food and Drug Administration.4,5
Evidence for clinical benefit of docetaxel in mCRPC
The TAX 327 study (n = 1,006) demonstrated an improvement in median OS with docetaxel every 3 weeks plus prednisone compared to mitoxantrone plus prednisone: 18.9 months vs. 16.5 months (hazard ratio [HR]: 0.76; 95% confidence interval [CI]: 0.62–0.94; P = 0.009). 4 Notably, patients receiving weekly docetaxel did not exhibit a survival extension compared to mitoxantrone. The most common toxicities of the every 3–week docetaxel arm were fatigue (53%), alopecia (65%), neutropenia (32%), and neuropathy (30%), although febrile neutropenia was uncommon (3%). 4 A second landmark trial, the SWOG-9916 trial, also demonstrated improved survival for docetaxel plus estramustine compared to mitoxantrone plus prednisone, but this combination is not commonly used due to the gastrointestinal and cardiovascular toxicities of estramustine. 5 Notably, analyses of both of these trials also demonstrated that PSA decline ≥30% within 3 months was a moderate surrogate for improved survival.25,26
Intermittent docetaxel treatment may be a reasonable strategy. While the above landmark Phase III trials aimed to deliver 10–12 cycles of docetaxel, in the ASCENT study (n = 250), patients who achieved a PSA ≤4 ng/mL could choose a chemotherapy holiday. 11 Treatment was resumed when the PSA increased by ≥50% and was ≥2 ng/mL, or if there was other evidence for disease progression. The median duration of the treatment holiday was 18 weeks (range: 4–70 weeks) and 45.5% of patients exhibited a ≥50% PSA decline following the second course of treatment after the holiday. 27
The combination of docetaxel with carboplatin yielded ≥50% PSA declines in ~20% of patients as second-line chemotherapy in mCRPC progressing after prior docetaxel-based chemotherapy (n = 34).28,29 However, owing to the lack of randomized trials, it is unclear if the addition of platinums confers a survival impact.
Benefit of docetaxel in castration-sensitive disease
Recently, randomized Phase III trials have validated the impact of docetaxel in metastatic castration-sensitive prostate cancer (mCSPC). The CHAARTED trial (n = 790) demonstrated a relatively large extension of median OS by combining six cycles of docetaxel with ADT (44.0 months vs. 57.6 months; HR: 0.61; P < 0.001). 30 Those with extensive metastatic disease (who constituted 65% of all patients), defined as visceral disease or ≥ bone lesions with ≥1 lesion outside the spine and pelvis, exhibited an even larger increment in median OS, from 32.2 months to 49.2 months. Then, the large STAMPEDE trial comparing 1,184 patients receiving ADT vs. 1,185 patients receiving ADT plus docetaxel demonstrated an increased median OS from 67 months to 77 months by adding six cycles of docetaxel plus prednisolone to ADT (HR: 0.76; P = 0.003).31,32 In contrast, nine cycles of docetaxel combined with ADT did not demonstrate improved OS in the smaller Phase III GETUG-AFU-15 trial, although progression-free survival (PFS) was prolonged. 33 Finally, early data indicate a potential benefit for the addition of docetaxel to ADT in patients with high-risk nonmetastatic disease.34,35 The GETUG-12 Phase III trial (n = 413) combined four cycles of docetaxel plus estramustine with ADT plus local therapy and demonstrated improved relapse-free survival (HR: 0.71; P = 0.017). Similarly, the Phase III RTOG-0521 trial (n = 563) demonstrated improved 4–year survival (89% vs. 93%; HR: 0.70; P = 0.04) with the addition of six cycles of docetaxel plus prednisone to ADT plus radiation therapy. 35 Overall, the larger increments of OS with docetaxel in CSPC suggest lower intrinsic resistance to docetaxel in this setting compared to the CRPC setting. Furthermore, patients exposed to docetaxel in the castration-sensitive setting may potentially be relatively docetaxel-resistant when the disease progresses to the castration-resistant state, due to the biology of the disease as well as partly due to acquired resistance induced by prior docetaxel exposure.
Cabazitaxel
Mechanism of action
Cabazitaxel (Jevtana®; Sanofi-Aventis) is a semisynthetic taxane drug that inhibits micro-tubule disassembly and displays antineoplastic activity in cell lines with p-glycoprotein overexpression, which is also a proposed mechanism for docetaxel resistance.36,37 Moreover, cabazitaxel can penetrate the blood-brain barrier in preclinical systems and can also inhibit nuclear AR transport. 38
Clinical evidence for efficacy of cabazitaxel in mCRPC
Activity of cabazitaxel in docetaxel-refractory mCRPC was shown in the Phase III TROPIC trial. 19 A total of 755 patients with mCRPC who had progressed, a median of ~4 weeks after the previous cycle of docetaxel, were randomized to prednisone 10 mg with mitoxantrone 12 mg/m2 or cabazitaxel 25 mg/m2 every 3 weeks for a maximum of 10 cycles. The trial showed improvement in median OS of 2.4 months for cabazitaxel over mitoxantrone (15.1 months vs. 12.7 months; HR: 0.70; P = 0.0001). There was also improvement in PFS, PSA response, and time to tumor progression. 19 This led to the approval of the drug in 2010 in the postdocetaxel setting. Cabazitaxel was associated with substantial myelosuppression, including toxic deaths in ~5% of patients. Prophylactic granulocyte colony-stimulating factor (G-CSF) support in selected patients at risk for neutropenic sepsis appears to improve the safety of this regimen.39–42 Indeed, with preemptive use of G-CSF when indicated based on performance status, comorbidities, and age, cabazitaxel was well tolerated and associated with quality-of-life benefits, low incidence of neuropathy, and no toxic deaths.
Recently, two postmarketing Phase III studies were reported: FIRSTANA (NCT01308567) compared doce-taxel plus prednisone vs. cabazitaxel 25 mg/m2 or 20 mg/m2 plus prednisone as first-line chemotherapy for mCRPC with the objective of demonstrating superiority for OS with cabazitaxel; and PROSELICA (NCT01308580) compared two doses of cabazitaxel (20 mg/m2 vs. 25 mg/m2) as postdocetaxel chemotherapy for mCRPC with the objective of demonstrating noninferiority of the lower dose.43,44 FIRSTANA did not demonstrate superiority of both doses of cabazitaxel over conventional docetaxel, with a median OS of ~2 years for all arms, although objective measurable tumor responses were higher with cabazitaxel 25 mg/m2 compared to doce-taxel (41.6% vs. 30.9%, P = 0.0370). There were interesting differences in the toxicity profile: grade 3–4 adverse events occurred in 41.2%, 60.1%, and 46.0% of patients with cabazitaxel 25 mg/m2, cabazitaxel 20 mg/m2, and docetaxel. Neutropenic fevers, diarrhea, and hematuria were more common with cabazitaxel 25 mg/m2, while peripheral neuropathy, peripheral edema, alopecia, and nail disorders were more common with docetaxel. PROSELICA confirmed the noninferiority for OS (~14–month median survival for both doses) of cabazitaxel at a dose of 20 mg/m2 compared to 25 mg/m2. Although PSA and Response Evaluation Criteria in Solid Tumors (RECIST) response rates were higher with cabazitaxel 25 mg/m2, grade 3–4 adverse events were also higher with this dose: 54.5% vs. 39.7%. These data suggest that 20 mg/m2 should be preferred as the more optimal dose in most circumstances, and additionally, this may be hypothesized to be more cost-efficacious. Moreover, TAXYNERGY, a randomized Phase II trial evaluated an early switch in taxane (docetaxel to cabazitaxel or cabazitaxel to docetaxel) in patients with decline in PSA <30% within 3 months. 45 Overall, 35 of 63 (55.6%) patients exhibited PSA response, which appeared to exceed the historical response rate of ~45% when using docetaxel.
A phase II trial examined the role of carboplatin in combination with cabazitaxel and granulocyte growth factor support for the “anaplastic” or aggressive variant prostate cancer by clinical criteria defined previously. It showed additional benefit from a platinum–taxane combination.46–48 These previously described anaplastic criteria were exclusive visceral or predominantly lytic bone metastases, bulky tumor masses, low PSA levels relative to tumor burden, or short response to prior ADT. The median PFS was 3.8 months with cabazitaxel vs. 5.7 months with the combination of cabazitaxel and carboplatin (P = 0.009). 43 PSA and objective response rates also improved with combination therapy. Thus, further investigation of this platinum–taxane chemotherapy doublet may be warranted for the anaplastic variant subgroup.
Clinical Evidence of Resistance and Cross-Resistance with Docetaxel and Cabazitaxel
Although newer chemotherapeutic agents have shown some survival benefit, the improvement is modest at best.13,19 The issue of acquired resistance or de novo resistance to chemotherapeutics has posed a challenge. Cross-resistance can also occur between docetaxel and cabazitaxel, between taxanes and androgen-targeting agents, and between androgen-targeting agents.
The PSA response rate is only ~25% for abiraterone acetate following enzalutamide or the reverse sequence, and radiographic responses are rare. In a retrospective study of patients receiving enzalutamide after abiraterone, PSA response rate (22% vs. 26%; P = 0.8), median time to radiologic/clinical progression (4.6 months vs. 6.6 months; P = 0.6), and median OS (10.6 months vs. 8.6 months; P = 0.2) did not differ significantly between docetaxel-treated and docetaxel-naive patients. 49 Enzalutamide also induced modest PSA responses and median survival of only 8.3 months in patients progressing following both chemotherapy and abiraterone. 50 In another report, treatment with either enzalutamide or docetaxel following abiraterone produced modest PSA responses and median PFS of only ~4.5 months.51,52 The activity of abiraterone acetate following enzalutamide and docetaxel also is modest.53,54
Interestingly, cabazitaxel appeared to retain moderate activity following docetaxel and novel antiandrogen drugs in one retrospective study. 55 In this retrospective study, 79 patients who had progressive mCRPC after docetaxel and abiraterone acetate displayed benefit from cabazitaxel. PSA decline ≥30% occurred in 48 patients (62%), and the median PFS and OS were 4.4 months and 10.9 months, respectively. In another study, cabazitaxel appeared active when given after both abiraterone and/or enzalutamide. 56 In this retrospective study of 41 patients, ≥50% PSA declines were seen in 16 of 41 (39%) patients, and objective radiologic responses occurred in three of 22 (14%) evaluable patients.
Mechanisms of Resistance to Chemotherapy
Resistance to chemotherapy can be attributed to specific mechanisms intrinsic to prostate cancer biology or general mechanisms common to different tumor types or drug pharmacokinetics (Fig. 1). 57

Pathways of chemotherapy resistance in metastatic castration-resistant prostate cancer.
Continued androgen–AR signaling
Activation of AR by androgens not only stimulates proliferation but also inhibits apoptosis of prostate cancer cells, leading to tumor growth and progression. Increased AR expression, AR gene amplification, mutations, alterations in coregulators, and continuous production of androgens within the tumor tissue and adrenal glands owing to the activity of cytochrome P450 (CYP)-17, among other enzymes, may engender continued activity of the androgen axis despite castrate serum testosterone levels, which may fuel tumor growth and resistance.58–60 Additionally, nonandrogen molecules, such as estrogens, progestin, and other oncogenic signaling molecules, may bind the promiscuous altered AR.61,62 Signal transduction pathways, cross talk between AR and human epidermal growth factor receptor (HER)-2/3 receptors, SRC family of kinases, transforming growth factor (TGF)-β are all implicated in the activation of AR even in the absence of androgens. 57 AR, when present in cytoplasm, is bound to heat shock proteins (HSPs) such as HSP-90 in both normal and prostate cancer cells. Testosterone is converted to dihydrotestosterone, which causes conformational change by dimerization and phosphorylation of the receptor, which dissociates AR from HSP. This change causes trafficking of AR to the nucleus, mediated by dynein, where it binds along with the coactivators and corepressors such as FOXO1 to the androgen response elements of DNA in the gene promoter and enhancer regions and induces transcriptional activation of the target growth-promoting genes.3,21–23 Indeed, the survival increments conferred by enzalutamide and abiraterone acetate following docetaxel exposure attest to the continued relevance of signaling via the androgen–AR axis in the late postdocetaxel phase of the disease.
Interestingly, recent retrospective studies suggest that the AR splice variant, AR-v7 and circulating free (cf)-DNA alterations of AR are associated with resistance to both abiraterone acetate and enzalutamide. 63 In contrast, hypothesis-generating studies suggest that tumors harboring AR-v7 may continue to respond to taxanes, while the TMPRSS2-ERG translocation may be associated with poor response to taxanes.64–66 One retrospective study showed that taxanes are associated with improved survival vs. antiandrogen drugs in AR-v7-expressing patients. 67 Additionally, conversion of AR-v7–positive to AR-v7–negative status appears to occur more frequently with taxanes than with antiandrogen drugs. 68
Upregulation of prosurvival cellular pathways
Docetaxel, in addition to stabilizing microtubules, also induces apoptosis by downregulating antiapoptotic proteins. 69 BCL2 expression was noted to be an independent predictor of survival in patients treated with taxanes.70,71 BCL2 inhibits mitochondrial release of cytochrome c and subsequently blocks the caspase cascade, thereby inhibiting apoptosis. During treatment with taxanes, BCL2 is phosphorylated, which prevents heterodimerization with other BCL family genes, thereby promoting apoptosis. Inherited resistance is noted in prostate cancer cells that do not express BCL2, indicating that taxanes’ mechanism of action relies at least partly on BCL2 inhibition. 70 Mcl1 (myeloid cell leukemia differentiation protein 1) and other members of the BCL family, such as BCL-xl (B-cell lymphoma-extra-large), are also involved in resistance to Interleukin (IL)-6, stromal cell derived factor-1, and cytokine-induced apoptosis. 71 Clusterin is a small heat shock glycoprotein overexpressed in most of the solid tumors, which promotes apoptosis by binding to various molecules such as BAX (BCL2-associated X protein) 72 and signal transducer and activator of transcription (STAT)–1. BAX and STAT are overexpressed and regulate clusterin expression in docetaxel-treated patients, indicating their role in cytoprotection from chemotherapy. 73
Inhibitors of apoptosis proteins (IAPs), mainly survivin and X-linked inhibitor of apoptosis (XIAP) prevent the processing of procaspase 3 to caspase 3, thereby inhibiting apoptosis. 74 Nuclear factor (NF)-κB plays a pivotal role in mounting an inflammatory response. Translocation of NF-κB leads to activation of the genes for IL-6, stress response elements, and many antiapoptotic elements such as IAPs.75,76 Tumor necrosis factor (TNF)–α inhibits apoptosis by activating NF-κB and its downstream pathway, including IL6 and IL8, in androgen-independent prostate cancer cells, whereas it promotes apoptosis in androgen-dependent cancer cells. 77 IL-8 acts through chemokine receptors 1 and 2 (CXCR1 and 2) and is involved in promoting angiogenesis through overexpression of vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF).78,79
Aberrations of AR, erythroblast transformation-specific (ETS) genes, Tumor protein 53 (TP53), and Phosphatase and tensin homolog (PTEN) occurred in 40%–60% of 150 mCRPC cases in a recent study. Additionally, alterations of phosphoinositide-3-kinase, catalytic, alpha polypeptide A/B (PI3KCA A/B), R-spondin, RAF/ Rapidly Accelerated Fibrosarcoma1 (BRAF/RAF1), Adenomatous polyposis coli (APC), β-catenin, Zinc finger and BTB domain-containing protein 16/ Promyelocytic leukemia zinc finger protein (ZBTB16/PLZF), Breast cancer 2 (BRCA2), Breast cancer 1 (BRCA1), Speckle Type POZ Protein (SPOP), and Ataxia telangiectasia mutated (ATM) were seen. Interestingly, 23% harbored DNA repair pathway aberrations, which appear to correlate with responses to the polyadenosine diphosphate-ribose polymerase (PARP) inhibitor, olaparib, in a recent trial.80,81
Activation of PI3K (phosphoinositide 3 kinase), protein kinase B (PKB), and mammalian target of rapamycin (mTOR) pathways may lead to chemotherapy resistance by either upregulating multidrug resistance protein or by overexpression of various protooncogenes and growth factors such as cyclin D1, VEGF, and c-myc.82,83 Drugs inhibiting pathways such as the Hedgehog, β-catenin, epidermal growth factor receptor (EGFR), endothelin, mitogen-activated protein kinases (MAPK) pathways are also implicated in reviving sensitivity to chemotherapy in chemoresistant cell lines.84–86 The development of neuroendocrine prostate cancer (including small cell cancer) also appears to confer resistance, and a genomic signature correlating with neuroendocrine transformation has been identified. 87
Role of tumor microenvironment: angiogenesis and immune mechanisms
The tumor microenvironment plays a substantial role in cancer cell survival and development of resistance to chemotherapy. The majority of solid tumors are composed of tumor cells mixed with noncancerous cells supported by a disorganized vascular network. 88 The blood vessels are farther than in normal tissue, which lead to regions of hypoxia and impaired delivery of nutrients as well as improper clearance of metabolic breakdown products. The chaotic blood supply also limits the delivery of cytotoxic drugs including taxanes to the cancer cells. 88 The accumulation of metabolic by-products, including lactic acid, increases the acidity of the tumor environment, which also influences the drug uptake by tumor cells. 89 The tumor hypoxia may retard tumor proliferation, rendering them more resistant to cell cycle-active chemotherapeutics as well as promoting a more malignant phenotype. 90 The hypoxic state also leads to upregulation of genes that promote cell survival including hypoxia–inducible factor (HIF)-1. This inturn leads to suppression of apoptosis, increased receptor tyrosine kinase signaling and increased angiogenesis, thereby promoting cell survival and metastases. 91 The intratumoral drug uptake is impeded by high interstitial fluid pressure and absence of lymphatic flow, causing stasis of cytotoxic drugs with in the blood vessels. 92 The integrin receptors present on the cancer cells promote adhesion of cancer cells to extracellular matrix to form multidimensional spheres, thereby worsening drug delivery to tumor cells. 89
As previously discussed, chemotherapy resistance can occur via overexpression of growth factors and cytokines such as IL-6 and NF-κB produced in the tumor stroma. The TGF-β, FGF, β-catenin, and mTOR pathways, in conjunction with the hypoxic state, are involved in the development of epithelial–mesenchymal transition (EMT).93,94 Indeed, markers of EMT have been strongly associated with docetaxel resistance in preclinical studies. 95 This process not only is involved in promoting invasiveness and chemotherapy resistance but also has been linked to development of metastases. 96
Additionally, prostate cancer cells secrete various cytokines, such as TGF, VEGF, endothelin-1, FGF, and bone morphogenetic protein, which can influence bone homeostasis either by modifying growth factors present in the osseous microenvironment or exerting direct effect on the osteoblast. 97 This leads to osteoblast as well as tumor cell proliferation. Chemokine ligand-2 stimulates tumor cells and osteoclasts, thus promoting bone metastases. 98 This paracrine secretion of cytokines may be induced by treatment with chemotherapy, indicating the role of chemokine ligand-2 in chemoresistance.
Drug efflux pump
The multidrug-resistant phenotype, mediated by the ATP-dependent drug efflux pump p-glycoprotein, appears central in the mechanism of chemotherapy resistance. 99 Multidrug resistance proteins (MDRPs) belong to ATP-binding cassette transporters and include p-glycoprotein and ABCB4 (encoded by MDR2 gene), and ABCC1 (encoded by MRP1 gene); they act as drug efflux pumps for a variety of chemotherapy agents, including taxanes. 100 P-glycoprotein is encoded by the multidrug resistance -1 (MDR1) gene, which is upregulated in cancer cells treated with docetaxel. 101
Microtubule alterations
Structural or functional alterations in the microtubules targeted by taxanes provide an additional mechanism of resistance. Upregulation of β-tubulin isotypes III and IV, or β-tubulin mutations that affect docetaxel binding, or posttranslational modifications in β-tubulin that confer the structural changes in microtubules may lead to taxane resistance.102–104 Functional changes such as alterations in the binding site (promoting β-tubulin detyrosination), microtentacles (that enhance endothelial engagement), alterations in γ-actin, and TXTR1–mediated thrombospondin repression may also contribute to taxane resistance.105,106
Potential Role of Novel Agents and combinations in Overcoming chemoresistance
Combinations of chemotherapy and antiandrogen drugs
A multipronged strategy is necessary to overcome chemoresistance. One rational strategy is to investigate combinations of chemotherapy with antiandrogen drugs, because both classes of agents are known to yield survival benefits as single agents. Phase I trials have already demonstrated the feasibility of combining cabazitaxel 25 mg/m2 with abiraterone-prednisone. 107 However, in the trial combining cabazitaxel 25 mg/m2 and abiraterone-prednisone, seven patients (25.9%) required dose reduction of cabazitaxel due to toxicities, but all patients received ≥80% of the planned dose intensity. 107 The combination of cabazitaxel 25 mg/m2 (with routine PEG-G-CSF) and enzalutamide is being evaluated in a Phase I trial (NCT02522715). Notably, the combination of enzalutamide and docetaxel is feasible and safe; indeed, the PRESIDE Phase III trial is comparing continued enzalutamide plus docetaxel vs. docetaxel after disease progression on enzalutamide alone. 108 Furthermore, enzalutamide, abiraterone, and prednisone were combined in full single-agent doses, and Phase III evaluation of this combination is ongoing. 109 No pharmacokinetic interactions have been seen, and toxicities have been manageable with all of these combinations. The combination of prednisone, abiraterone, cabazitaxel, and enzalutamide as first-line therapy for mCRPC will be evaluated in a planned Phase I trial. This trial will allow prior docetaxel for castration-sensitive disease.
Combination of chemotherapy with novel biologic agents
The combination of docetaxel with novel agents has heretofore been unsuccessful in unselected patients. Phase III trials attempting to build on a template of docetaxel by combining with bevacizumab, aflibercept, DN101 (vitamin D analog), GVAX, dasatinib, atrasentan, lenalidomide, and custirsen have all been disappointing with no increments.110–115 Additionally, compromising the delivery of docetaxel owing to the added toxicities of combinations appeared to harm survival. 114 Hence, the development of docetaxel plus biologic agent combinations must proceed cautiously and be guided by rational patient selection. An ongoing Phase III trial is evaluating the role of combining DCVAC, an autologous dendritic cell-based vaccine (pulsed with killed prostate cancer cell lines), with docetaxel-based chemotherapy as first-line therapy for mCRPC.
Novel agents for single-agent therapy
Novel agents may also be active as single agents following progression on chemotherapy and some of the ongoing trials in the chemoresistant setting have been outlined in Table 1. PARP inhibitors appear particularly promising in this regard. A Phase II trial enrolling 50 patients with mCRPC were treated with olaparib. 81 These patients were heavily pretreated and all had received prior docetaxel, 49 had received abiraterone or enzalutamide, and 29 had received cabazitaxel. Sixteen of 49 (33%) evaluable patients had a response defined by measurable tumor regression, ≥50% PSA decline, or reduction in circulating tumor cell count. Of the 16 patients with alterations in DNA repair genes - BRCA1/2, ATM, Fanconi's anemia genes, and CHEK2 - 14 (88%) had a response to olaparib, including all seven patients with BRCA2 loss and four of five with ATM aberrations. Toxicities were manageable and consistent with previous olaparaib trials. A phase III trial is being planned.
Ongoing trials evaluating new agents and combinations to overcome chemoresistance.
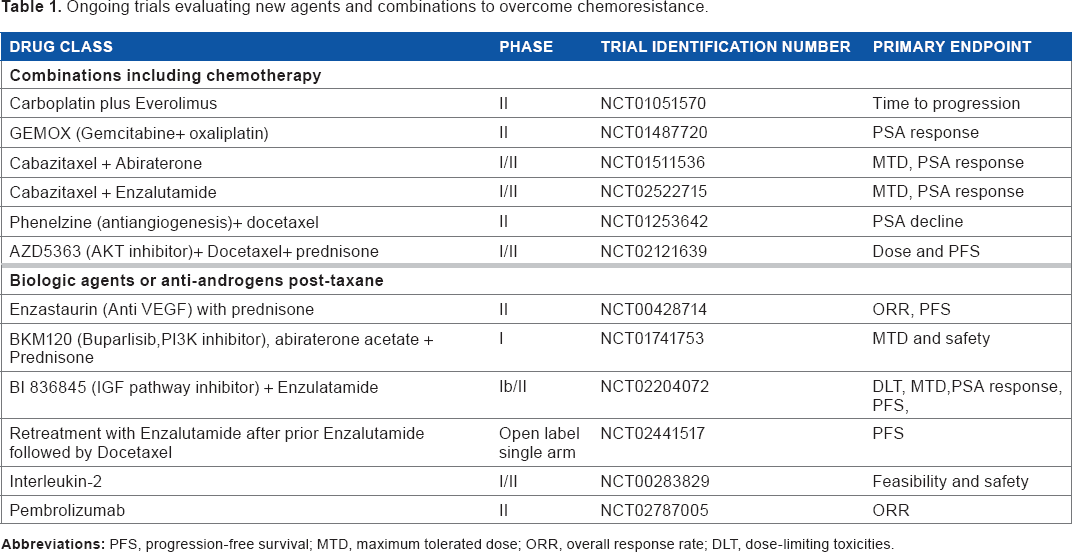
BTK120 (Bruton tyrosine kinase 120), an inhibitor currently in open-label, single-arm Phase II clinical trial (NCT01385293) for mCRPC, progressed on previous therapies including cytotoxic agents. BI-836845 is a human monoclonal immunoglobulin (Ig)-G lambda antibody against insulin-like growth factor (IGF)-1 and IGF2, which is implicated in inhibiting apoptosis. In a Phase Ib/II trial, BI-836845 in combination with enzalutamide is being compared with enzalutamide alone in patients who have progressed on docetaxel and abiraterone. Monoamine oxidase A (MAO-A) is expressed commonly in high-grade prostate cancer and is implicated in tumor survival and metastases by EMT and angiogenesis. Phenelzine is a MAO-A inhibitor and is being studied in a Phase II trial in combination with docetaxel in patients who have progressed on docetaxel. One randomized Phase II trial is evaluating ADT with or without palbociclib, a cyclin-dependent kinase 4/6 inhibitor, for mCSPC and intact Retinoblastoma (RB). Palbociclib may potentially warrant evaluation even in postchemotherapy patients, considering the continued relevance of dependence on the androgen axis and resistance to antiandrogen drugs even after chemotherapy. The AURKA gene appears to be involved in neuroendocrine transformation, suggesting that aurora kinase inhibitors, such as MLN8237, may be active in this setting.116,117
Several novel second- and third-generation antiandrogen agents are undergoing Phase III evaluation (eg, ODM201, ARN509, galeterone, and VT464), although these trials do not specifically address chemoresistance because these trials have enrolled chemotherapy-naive patients. An extensive discussion of these agents is therefore beyond the scope of this review.
Immunotherapy was dealt a setback in the context of mCRPC when two Phase III trials evaluating ipilimumab, a cytotoxic T-lymphocyte antigen (CTLA)-4 inhibitor, in the pre- and post-docetaxel settings did not extend OS. 118 Moreover, programmed death (PD)-1 inhibitors preliminarily do not appear to have robust activity in unselected patients with advanced prostate cancer. 119 Interestingly, sipuleucel-T upregulates PD-1–expressing T-cells when administered as a neoadjuvant therapy for localized disease and preliminary evidence for clinical activity of pembrolizumab in enzalutamide-resistant patients, suggesting a role for PD-1 inhibitors in selected patients.120,121 Indeed, one phase II trial is evaluating pembrolizumab in men with tumor PD-L1 expressing or non-expressing mCRPC previously treated with docetaxel (Keynote-199). Additionally, targeting angiogenesis has not yielded benefits using either the combination strategy with docetaxel (bevacizumab and aflibercept) or postdocetaxel therapy with sunitinib plus prednisone or cabozantinib.110,122–124
Conclusions and Future Directions
The approval of docetaxel chemotherapy in prostate cancer treatment ushered in an era in which improvement in OS became a well-defined, achievable end point. However, resistance still occurs, and responses are limited in men with mCRPC. There is a continuing need to understand varying mechanisms of resistance and ways to overcome them.
In order to better understand tumor biology and mechanisms of chemoresistance, pre- and postchemotherapy molecular analyses of tumor genomic material are critical. Additionally, the mechanisms of resistance to chemotherapy following administration of other classes of agents, such as anti-androgen agents, may also be applicable considering the cross-resistance between them. Rational selection of patients is also important to produce large increments and to develop precision medicine.
Author Contributions
Conceived and designed the experiments: GS, VL, JA. Analyzed the data: GS, VL, JA. Wrote the first draft of the manuscript: VL. Contributed to the writing of the manuscript: GS, VL, JA. Agree with manuscript results and conclusions: GS, VL, JA. Jointly developed the structure and arguments for the paper: GS, VL, JA. Made critical revisions and approved final version: GS, VL, JA. All authors reviewed and approved of the final manuscript.