
Abstract
Ipilimumab is a monoclonal antibody directed against CTLA4 T-lymphocyte antigen used as cancer therapy. Immune-related adverse events are common side effects and may include hypophysitis-related hypopituitarism. The clinical features of six patients with ipilimumab-induced hypophysitis (IH) are described. The clinical features of IH reported in clinical trials, including the incidence of IH by gender and the likelihood of adrenal axis recovery, are summarized. Following the development of IH, most patients remain on glucocorticoid replacement despite efforts to withdraw therapy. Analysis of gender information in published clinical trials suggests that men are more prone to developing IH than women, and few patients fully recover the pituitary-adrenal axis function. Ipilimumab and other drugs within its class are likely to be used to treat many forms of cancer. Endocrinologists should anticipate a significant increase in the incidence of autoimmune hypophysitis. Strategies for early detection of IH and long-term management should be considered.
Introduction
Drug-induced autoimmune hypophysitis is occurring more frequently as a result of increasing use of immune checkpoint inhibitors for various malignancies, including melanoma and lung and prostate cancers. This class of therapy will likely be used increasingly to treat other forms of cancer in the future. Thus, we should expect to see hypophysitis more often in older patients with multiple comorbidities.
Monoclonal antibodies against cytotoxic lymphocyte antigen 4 (CTLA4), ipilimumab in particular, inhibit the CTLA4-mediated inhibition of T lymphocytes, leading to an immune-mediated antitumor response (Fig. 1). Ipilimumab is now an approved therapy for metastatic melanoma because significant objective responses and overall survival benefit have been demonstrated with its use. 1 Immune-related adverse events (IRAEs) are recognized complications of this treatment. The most commonly reported IRAEs are enterocolitis, skin rash (including vitiligo), and hepatitis. The most common endocrinopathy associated with ipilimumab use appears to be hypophysitis with hypopituitarism, which has been reported in 0%–17%, followed by hypo- and hyperthyroidism secondary to thyroiditis in 2.7% and 0.3%, respectively, and primary adrenal insufficiency in 2.1%. 1

Ipilimumab's mechanism of action. CD28 (T-lymphocyte costimulatory receptor) and CTLA4 (T lymphocyte coinhibitory receptor) have a common tumor antigen ligand (B7). CTLA4, when expressed on T-cell membrane, has a higher affinity for B7 binding, which leads to cancer immune tolerance. Ipilimumab blocks CTLA4 and leads to anticancer immune effects through T-lymphocyte activation.
The mechanism by which hypophysitis occurs after iplimumab exposure is not clear. The leading hypothesis is T-lymphocyte-mediated pituitary destruction secondary to immune system activation. 2 The anterior pituitary is always affected by ipilimumab-induced hypophysitis (IH). Posterior pituitary dysfunction after ipilimumab therapy in the form of diabetes insipidus has been reported in only one case. 3
Description of Patients
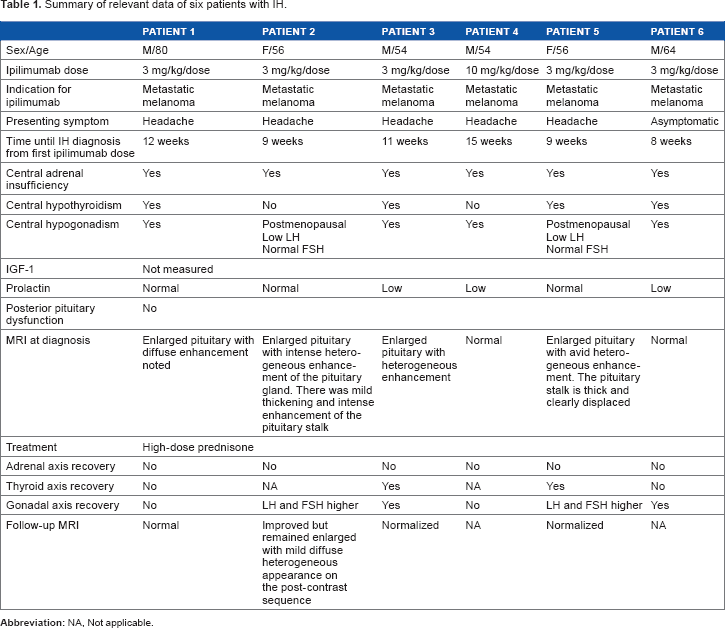
Table 1 summarizes relevant clinical data and clinical course of six patients with IH seen at our academic medical center. The Ottawa Health Science Network Research Ethics Board exempted this review of cases from ethics board approval, as the patients were seen by the authors in their clinical role. Diagnosis of hypophysitis was made when biochemical evidence of hypopituitarism was detected by screening blood work or after patients presented with symptoms suspicious of hyophysitis (mainly new-onset headache). Biochemical assessments for hypopituitarism included testing of adrenal, gonadal, and thyroid axes plus electrolyte and prolactin levels. Available hormonal levels are shown in Table 2. Central hypoadrenalism was diagnosed based on low morning or random (in the patients with new onset headache) cortisol with inappropriately normal or low adrenocorticotropic hormone (ACTH). Central hypothyroidism was defined as low free T4 with inappropriately normal or low thyroid stimulating hormone (TSH). Hypogonadotrophic hypogonadism was diagnosed in male patients based on low testosterone with inappropriately normal or low luteinizing hormone (LH) and/or follicle-stimulating hormone (FSH). The two female patients were postmenopausal and had lower LH and FSH at presentation compared to levels after treatment with high-dose glucocorticoid therapy. This suggested the presence of partial gonadotrophic cell dysfunction at the time of presentation. The follow-up periods ranged from 6 to 13 months. Insulinlike growth factor (IGF)-1 levels were not measured because a low IGF-1 may be inadequate to diagnose growth hormone (GH) deficiency in this population. Furthermore, low IGF-1 would not influence clinical management since GH replacement is contraindicated in the setting of metastatic cancer. Patient 6 was referred to us 8 weeks after diagnosis of IH by his oncologist, and his ACTH, LH, and FSH upon diagnosis were not available. None of the patients had symptoms or electrolyte abnormalities suggestive of diabetes insipidus. Four out of six patients had pituitary enlargement on magnetic resonance imaging (MRI) upon diagnosis (Fig. 2).
Summary of relevant data of six patients with IH.
Hormonal profiles of the patients upon diagnosis of IH and in follow-up@.


MRIs of patients with abnormal findings.
Discussion
Clinical Presentation of Ipilimumab-Induced Hypophysitis
IH has been described to occur as early as 4 weeks after first exposure to ipilimumab, but the median time interval between the first treatment and presentation of IH is 11 weeks.3,4 Headache is the most commonly reported presenting symptom. Other presenting symptoms described in the literature are largely nonspecific and include fatigue, weakness, confusion, hallucinations, memory loss, labile mood, insomnia, anorexia, temperature intolerance, chills, decreased libido, and erectile dysfunction.5,6 Visual impairment due to pituitary swelling has also been reported. 4 In general, the presenting symptoms of IH are similar to those of classic lymphocytic hypophysitis (LyH).
Biochemical Profile
In IH, both ACTH and TSH secretion are affected in 60%–100% of patients according to different cohorts.7–9 Hypogonadotrophic hypogonadism has been reported in 83%–87% of male patients. Min et al reported GH deficiency (based on low IGF-1 level) in three out of five patients who had their IGF-1 measured. 9 Posterior pituitary involvement appears to be much less common, with only a single case report describing diabetes insipidus after ipilimumab. 3 In one case report, a patient was described to have hyponatremia secondary to syndrome of inappropriate antidiuretic hormone (SIADH). Of note, this patient also had central adrenal insufficiency, which could have been the cause of hyponatremia, and highlights some of the challenges of diagnosis and the need for a screening protocol for patients at risk. 10 Serum prolactin is typically normal, but has been reported to be high or low in up to 25% of the patients.5,6
MRI Findings
MRI is an important investigational modality in this population to rule out sellar metastasis. Symmetrical enlargement of the pituitary gland is reported in 12%–88% of patients with IH in different cohorts. 4 MRI also usually shows homogeneous enhancement of the pituitary gland similar to LyH cases. 4
Pathogenesis of Ipilimumab-Induced Hypophysitis
The pathogenic mechanism of anti-CTLA4 monoclonal antibody-induced hypophysitis is largely unknown. Because the presenting clinical features, MRI findings, and hormone profiles resemble what is seen in classic LyH, and the immune checkpoint inhibitors effectively “unleash” the immune system, the pathogenic mechanism is suspected to be autoimmunity with lymphocytic destruction of pituitary cells. 2 This is supported by the fact that hypophysitis never occurs when ipilimumab is combined with cytotoxic chemotherapy, presumably due to lymphocyte depletion from cytotoxic agents. 11 Iplimumab is also capable of inducing autoantibodies. 12
Iwama et al have documented the development of pituitary autoantibodies in seven patients with IH who had negative antibodies prior ipilimumab treatment. In contrast, none of the 13 patients who did not develop IH developed pituitary autoantibodies. The antibodies recognized predominantly TSH-secreting cells and, less frequently, FSH- or ACTH-secreting cells. Subsequently, pituitary expression of CTLA4 was established on a murine model and in human pituitary gland. The CTLA4 expression was confined to prolactin and TSH-producing cells in the murine model. It was concluded that the trigger for IH is a type II hypersensitivity reaction, in which the CTLA-4 antibody binds to the cognate antigen expressed on pituitary cells (prolactin and TSH-producing cells), activates the complement, and initiates tissue destruction. It is not known whether CTLA4 antigen is expressed in posterior pituitary cells. Lack of expression could explain why posterior pituitary dysfunction is not common in patients with IH. Further investigations of pituitary's CTLA4 expression as well as distribution in humans and its implication in the pathogenesis of IH are required. 13
Obtaining pathological specimens that could confirm lymphocytic infiltration might be considered only in exceptional cases when high-dose glucocorticoid therapy fails to relieve compressive symptoms. To date, no pathologic examination of IH cases has been reported.
Development of autoimmune-adverse events such as hypophysitis might actually be favorable because the development of IRAEs correlated with better tumor response in some trials. 14
CTLA4 expression on T lymphocytes has been correlated with single nucleotide polymorphisms in the CTLA4 gene. In a Phase I trial of a CTLA4 blocker in patients with metastatic melanoma, Sanderson et al found that patients having a polymorphism associated with reduced CTLA4 expression had the lowest rate of melanoma recurrence. These patients also had a higher incidence of IRAEs. 15 IRAEs like hypophysitis can occur prior to the assessment of the response to therapy, so development of IH should be studied longitudinally to determine whether it could be a useful marker of treatment response. Further studies to clarify the association between CTLA4 gene polymorphisms and clinical responses to CTLA4 antibody therapy are needed.
Male Predominance of IH
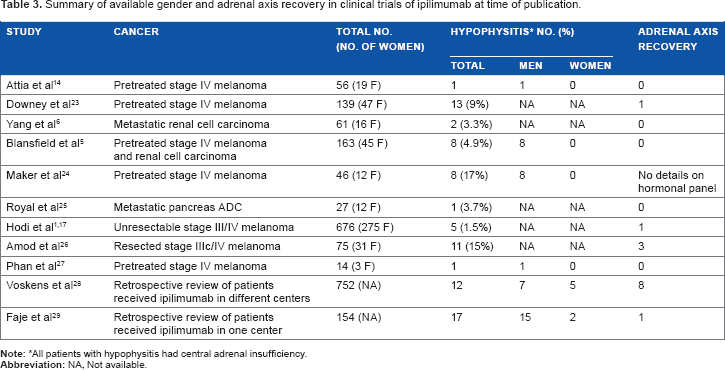
IH appears to preferentially affect men. This contrasts the findings of an epidemiologic review of patients with classic LyH, in which women were more frequently affected. However, the prevalence of LyH among the sexes varied depending on which pituitary segments were predominantly affected. The women to men ratio among the patients diagnosed was 6:1 for adenohypophysitis, 1:1 for infundibuloneurohypophysitis, and 1.9:1 for panhypophysitis. 16 Table 3 summarizes the data reported in ipilimumab clinical trials regarding the gender distribution of patients who developed IH. The higher incidence of IH in men has to be confirmed by further studies. The apparent higher incidence of IH in male patients could in part be due to the higher incidence of metastatic melanoma in men (male: female ratio, 2:1) and to the possible reporting bias since the sex of patients with IH is not reported in many of the published trials. Nonetheless, it seems that men are at least as susceptible as women to develop IH, unlike the female predominance of classic LyH.
Summary of available gender and adrenal axis recovery in clinical trials of ipilimumab at time of publication.
Recovery from Hypopituitarism after IH
Recovery of the hypothalamic–pituitary–thyroid axis has been reported in 37%–50% of patients4,9,17,18 and gonadal axis recovery occurred in 57% of men.4,16 Conversely, very few patients were able to discontinue glucocorticoid replacement due to persistent central adrenal insufficiency. Table 2 summarizes the published clinical trial data regarding pituitary–adrenal axis recovery in patients with IH.
Hypophysitis appears to be the only ipilimumab-induced IRAE that persists long after exposure to the drug. 5
Although prolonged high-dose glucocorticoid therapy used to treat IRAEs could lead to adrenal suppression, persistent central hypoadrenalism in most patients is probably due to the irreversible damage of corticotroph cells.
The older age and comorbidities of patients with IH are presumably associated with poor tolerance of relative adrenal insufficiency. This may make physicians reluctant to expose patients to prolonged glucocorticoid withdrawal protocols, which may in turn reduce the likelihood of recovery of the hypothalamic–pituitary–adrenal axis. Efforts should be made to minimize supraphysiologic glucocorticoid exposure to avoid dampening responses induced by therapies such as ipilimumab, which work by activating the immune system.
Other Immune Checkpoint Inhibitor Therapies and Hypophysitis
Tremelimumab is another CTLA4 blocker that has been investigated in cancer clinical trials, but it was found to be less effective than ipilimumab. The incidence of hypophysitis in tremelimumab-treated patients was reportedly 0.4%–2.5%.19,20 Whether the low rate of hypophysitis directly correlates with the lack of treatment response is unknown.
Monoclonal antibodies targeting programmed death-1 (PD-1), an inhibitory receptor expressed by T cells or one of its ligands (PD-1-L), are at early phases of development. Hypophysitis has been described in association with these agents, but with lower incidences.19,21,22
IH in Contrast to Classical Immune Hypophysitis
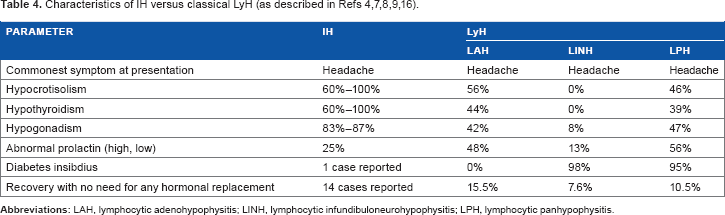
Populations that develop IH versus classical LyH are clearly different. Ipilimumab is an approved treatment for metastatic melanoma and under investigation for other metastatic cancers. Metastatic melanoma is more frequent in men, and the incidence increases with age. In contrast, classical LyH is typically a disease of young women, especially in the postpartum period. 16
Patrizio et al 16 did an extensive literature review for autoimmune hypophysitis and reported extensively on various epidemiologic aspects of the disease. Some of the epidemiologic findings of Patrizo et al are summarized in Table 4, contrasting them with what is known about IH. IH and classical LyH have a similar clinical presentation, with headache being the most common presenting symptom. IH predominantly affects the anterior pituitary lobe. Therefore, we chose to compare IH to lymphocytic adenohypophysitis (LAH) subtype of classical LyH. It appears that central hypothyroidism, central hypocortisolisma, and hypogonadotrophic hypogonadism are more common in patients with IH, while prolactin abnormalities are more common in patients with LAH. Up to 15.5% of patients with LAH recovered completely with no need for hormonal replacement, while only a few patients with IH have been reported to stop all the hormonal replacement due to persistent central hypocortisolism.
Conclusion
Clinicians should expect to see significant increases in the incidence and prevalence of hypopituitarism secondary to hypophysistis after immune checkpoint inhibitor (ipilimumab) therapy. Ipilimumab and drugs with similar mechanisms of action are likely to be used to treat many cancers, including lung, prostate, and metastatic melanoma.
Men treated with ipilimumab appear to develop IH as often, if not more often, than women, in contrast with the higher incidence of classic LHy observed in women. For all immune checkpoint inhibitors, determining the true incidence of hypophysitis is a challenge. Patients with metastatic cancer who will be considered for ipilimumab therapy are often older and have serious health issues. They may already be on thyroid hormone or glucocorticoids before they receive ipilimumab, and that may mask some of the hormonal changes that occur with IH. Symptoms of hypopituitarism could mistakenly be attributed to their comorbid conditions. Therefore, a high index of suspicion is required. Endocrinologists should collaborate with their local oncologists to develop systematic screening processes to ensure early detection of hypopituitarism because unrecognized adrenal insufficiency can be fatal. Patients with IH are likely to remain on long-term glucocorticoid replacement, putting them at risk of adverse effects associated with glucocorticoid treatment. Further research would be useful to determine whether the need for long-term glucocorticoid replacement is due to corticotroph destruction or is driven by the physicians' reluctance to expose patients to prolonged steroid-withdrawal protocols.
The presence of anti-pituitary antibodies as a potential causative mechanism for IH is possible but not confirmed. Further research to identify predisposing factors and elucidate the cascade of events that leads to hypophysitis is needed.
Prediction of response to therapy and risk of development of IRAEs should be explored. Investigating the utility of CTLA4 gene polymorphisms to predict response to CTLA blockers and risk of IRAEs should be pursued. Early identification of those at higher risk for IRAEs could be used for rational resource allocation with regard to screening and early diagnosis of IH in at risk patients.
Footnotes
Author Contributions
Wrote the first draft of the manuscript: MM. Contributed to the writing of the manuscript: MM, HL, DL, and AA. Agreed with manuscript results and conclusions: MM, HL, DL, and AA. Jointly developed the structure and arguments for the paper: MM and HL. Made critical revisions and approved final version: MM, DL, and HL. All authors reviewed and approved of the final manuscript.