
Abstract
The use of immune checkpoint inhibitors including ipilimumab and nivolumab has expanded for several tumors including melanoma brain metastasis. These have resulted in a growing spectrum of neurologic immune-related adverse events, including ones that are rare and difficult to diagnose and treat. Here, we present a patient with melanoma brain metastasis who was treated with immune checkpoint inhibitors and developed an Acute Motor Axonal Neuropathy. To our knowledge, this is the first case of Acute Motor Axonal Neuropathy as an immune-related adverse event associated with combination treatment of ipilimumab and nivolumab, who was successfully treated. A 28-year-old woman with metastatic BRAF V600E melanoma developed melanoma brain metastasis and was enrolled on Checkmate 204, a Phase 2 clinical trial using ipilimumab (3 mg/kg intravenous) and nivolumab (1 mg/kg intravenous) every 3 weeks for four cycles, followed by monotherapy with nivolumab (240 mg intravenous) every 2 weeks. A few days after Cycle 2 of ipilimumab and nivolumab, she developed a pure motor axonal neuropathy consistent with Acute Motor Axonal Neuropathy. She was treated with several immunosuppressive treatments including high dose methylprednisolone, immune globulin, and infliximab, and her motor neuropathy eventually improved several months after onset of symptoms. Unfortunately, she had progression of her systemic disease and died several months later. This is the first case reported of Acute Motor Axonal Neuropathy associated with ipilimumab and nivolumab, successfully treated with immune-suppressive therapy. As the field of immunotherapy expands with the increasing use of the immune checkpoint inhibitors, it is critical to increase our knowledge and understanding of the neurologic immune-related adverse events associated with immune checkpoint inhibitors. This includes the spectrum of rare neurologic immune-related adverse events, which can be quite difficult to recognize and treat. Early consultations with neurology may expedite a diagnosis and treatment plan in patients with unexplained weakness receiving immune checkpoint inhibitor therapy.
Introduction
Melanoma brain metastasis (MBM) represents a major cause of morbidity and mortality. Combination treatment with the immune checkpoint inhibitors (ICIs) ipilimumab (Ipi) and nivolumab (Nivo) was initially approved by the Federal Drug Administration (FDA) for the treatment of BRAF V600 wild-type (and soon thereafter expanded to include BRAF V600 mutated), unresectable melanoma based on its ability to demonstrate an increased response rate, prolonged response durations, and improvement in progression-free survival.1–4 However, a high number of immune-related adverse events (irAEs) has been associated with the use of ICIs including, rare and difficult to diagnose, neurological conditions. Here, we present a patient with MBM who was treated with Ipi and Nivo while enrolled on the Checkmate 204 clinical trial, 5 and developed Acute Motor Axonal Neuropathy (AMAN). To our knowledge, this is the first case reported of a patient diagnosed with AMAN associated with combination therapy of Ipi and Nivo, who was successfully treated with immune-suppressive therapy. We report this so that others may recognize this rare complication of ICI therapy, and immediately discontinue immunotherapy and initiate appropriate treatment.
Case report
A 28-year-old woman with widespread metastatic melanoma BRAF V600E mutant, involving the lungs, right breast, liver, and bone, was found to have a new asymptomatic metastatic lesion in her brain in the left temporal lobe, measuring approximately 0.5 cm, on magnetic resonance imaging (MRI; Figure 1). She was enrolled on the Checkmate 204 clinical trial, an open-label, multicenter, Phase 2 study to evaluate the intracranial clinical benefit in patients with metastatic melanoma and non-irradiated brain metastasis, treated with Ipi (3 mg/kg intravenous (IV)) and Nivo (1 mg/kg IV) every 3 weeks for four cycles, followed by monotherapy with Nivo (240 mg IV) every 2 weeks. 5 She tolerated well Cycles 1 (C1) and 2 (C2) of Ipi and Nivo, but she developed an acute and progressive diffuse motor weakness 9 days status post (SP) C2 (Figure 2, Supplementary Table 1). She had met criteria for a motor neuropathy Grade 3 and was removed from the study. 6
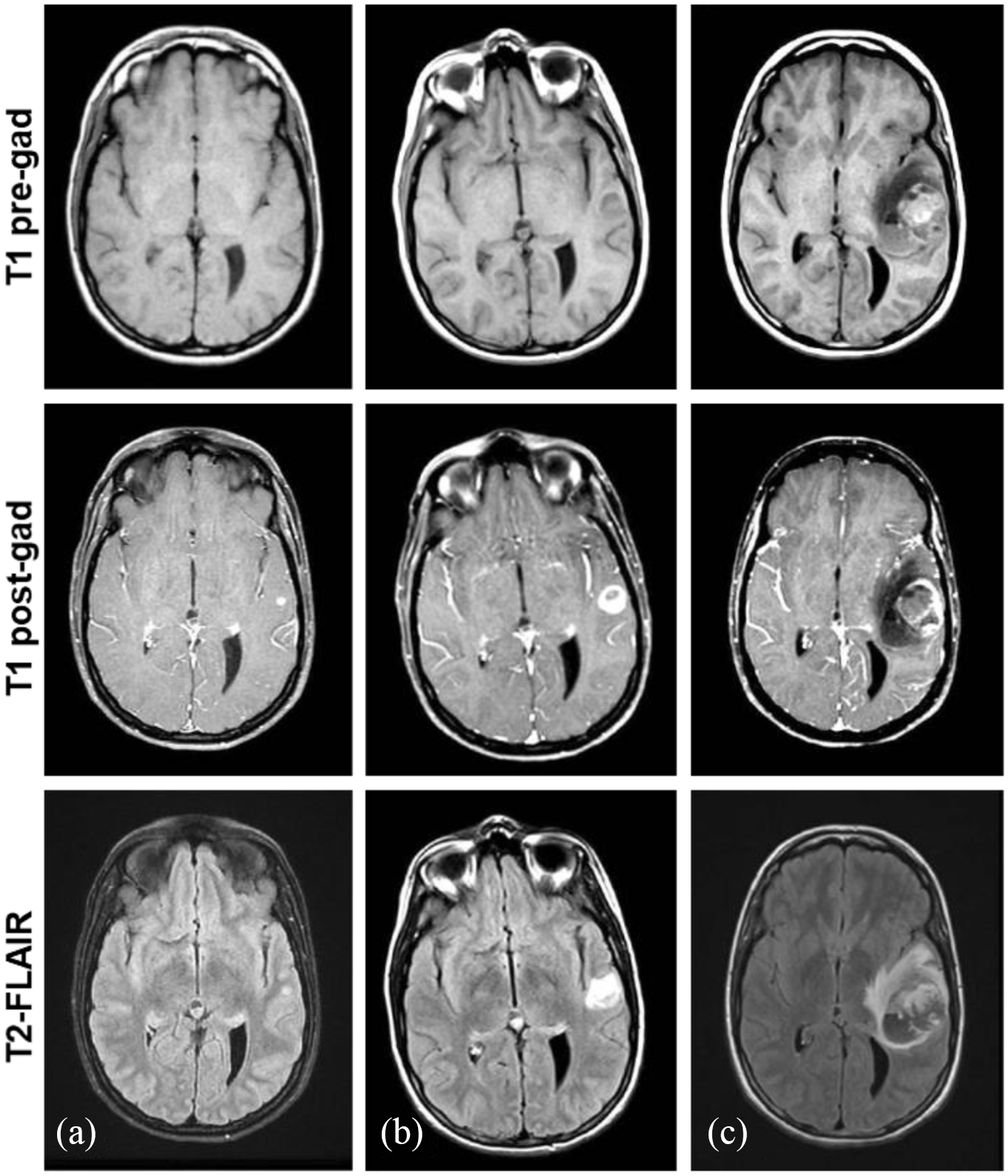
MRI brain with and without contrast shows progressive MBM. (a) Initial imaging revealed an asymptomatic solitary non-hemorrhagic lesion in the left temporal lobe measuring 0.5 cm in diameter. (b) Two months later, imaging showed an increase in the size of the lesion measuring 1.6 cm x 1.4 cm, with surrounding vasogenic edema on T2 fluid attenuated inversion recovery (FLAIR) sequences; but, no new metastatic lesions, acute hemorrhage or hypophysitis were identified. (c) Four months later, MRI revealed a large intracranial hemorrhage in the site of the known left temporal lobe lesion, measuring 4.7 cm x 3.4 cm with a 4 mm midline shift, with minimal enhancement suggesting the intracranial hemorrhage was related to treatment changes.
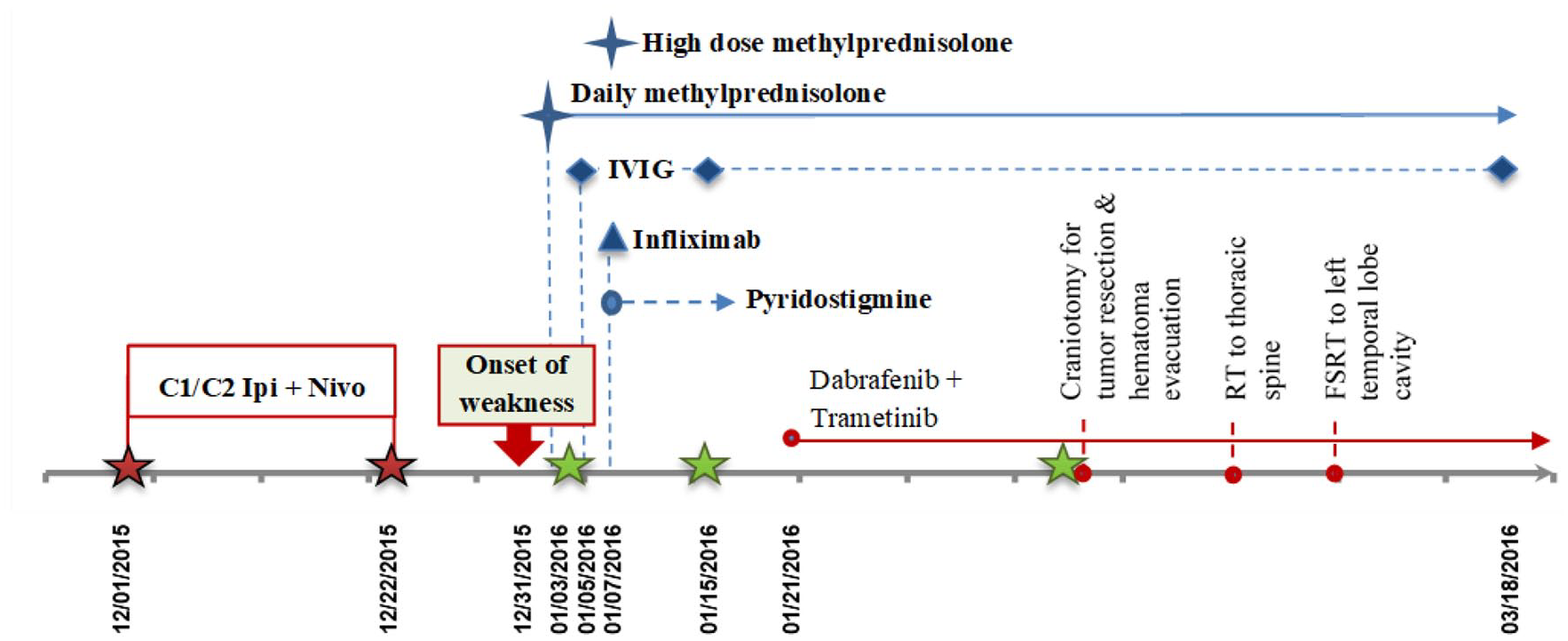
Treatment timeline.
Her initial examination was remarkable for a diffuse motor weakness that started 2 days prior presenting to the hospital, including neck flexion (2/5); bilateral (BL) deltoids (3/5), BL biceps (4–/5), BL triceps (3+/5); right (R) digit extension (2+/5), left (L; 3+/5); R hip flexion (1/5), L (2–/5); R knee extension (3–/5), L (3+/5); BL knee flexion (4–/5); R plantar and toe extension (2/5), L (3/5; see Supplementary Table 1). 7 Mental status, cranial nerves (CNs), and sensation to all modalities were normal. Deep tendon reflexes were normal on the L and reduced on the R, including her biceps, triceps, brachioradialis, and quadriceps. She had an ankle jerk on the L but not the R. She was immediately started on daily high dose IV methylprednisolone (IVMP) 1 mg/kg twice a day (BID; at 3D SP onset of symptoms; Figure 2). Despite steroids, her weakness continued to progress. At the next day, she had worse R shoulder flexion and BL iliopsoas muscles, and on the third day, developed a L facial nerve palsy with asymmetric L nasolabial fold and able to close the L eye but with diminished velocity, was unable to hold a note for more than 8 s, was weaker in BL deltoids, L digit extensors, but stronger L biceps and L quadriceps, and unable to obtain BL ankle jerks and quadriceps reflexes were difficult to obtain but present. She was started on IV immune globulin (IVIG; 0.4 g/kg daily for 5 days, at 5D SP onset of symptoms; Figure 2). On the fourth day of serial exams, she had a more pronounced L facial weakness, but was able to hold a note for 10 s with improved functional vital capacity (FVC). However, on Day 5 her weakness continued to progress despite IVMP and IVIG, and was treated with infliximab 5 mg/kg IV once, pyridostigmine 120 mg every 8 h (at 7D SP onset of symptoms), and a boost of high dose IVMP 1 g/day for 3 days (at 8D SP onset of symptoms; Figure 2). Her motor exam stabilized. She started to show some objective improvement on Day 9 of examination, at 21D SP C2 & 12D SP onset of symptoms (see Supplementary Table 1). She received a repeat cycle of IVIG at 15D and 77D given delayed signs of inflammation on repeat cerebrospinal fluid (CSF) analysis (Figure 2).
Regarding work up, the MRI of the brain demonstrated increase in the size of the lesion measuring 1.5 cm in diameter (three-fold increase in diameter compared with baseline) with some vasogenic edema that was not significant to cause her symptoms, and no other acute findings (Figure 1). Spine MRI showed widespread multilevel metastatic disease (image not shown) in the cervical, lumbar, and sacral vertebral bodies and an epidural lesion at T10 level, but no cord compression or evidence of leptomeningeal disease. Basic laboratory studies (i.e. complete blood count (CBC), complete metabolic panel (CMP), thyroid function panel, creatine kinase (CK), troponins) and serum paraneoplastic panel (i.e. antibodies (ABs) to neuronal nuclear (ANNA) Types 1, 2, 3; glial nuclear (AGNA) Type 1; purkinje cell cytoplasmic (PCA) Types 1, 1, Tr; amphiphysin; and CRMP-5-IgG) were unremarkable. ABs to acetylcholine (ACh) receptor (i.e. binding, modulating, and striational), voltage-gated calcium channel (VGCC), and ganglioside GM-1 (i.e. IgG and IgM normal titers) were also normal. A lumbar puncture revealed a normal opening pressure of 17.4 cmH2O, with CSF analysis remarkable for only mildly elevated white blood cell (WBC) count (8/mm3) with lymphocytic predominance (92%) and protein (73 mg/dL). CSF cytology was negative for malignancy. A repeat CSF analysis at 43D SP onset of symptoms was similar to initial with elevated WBC (59/mm3) and reactive lymphocytic pleocytosis, red blood cell (RBC; 98/mm3), and protein (200 mg/dL); in addition to elevated IgG index (1.0) indicating inflammation, but negative oligoclonal bands. A repeat complete paraneoplastic panel and ABs to GM1 were normal.
Further work up included electromyogram/nerve conduction studies (EMG/NCS) performed on three occasions (i.e. at 5D, 14D, and 44D SP onset of symptoms), which consistently revealed a pure motor axonal neuropathy, with significantly decreased compound muscle action potentials (CMAPs; i.e. median nerve CMAP of 0.417 mV, or 8.34% of the normal amplitude), normal conduction velocities (CVs; i.e. median nerve CV of 60 m/s), a reversible conduction block, and a lack of prolonged F waves (Figure 3(a)–(d), Supplementary Table 2). These studies also showed normal sensory nerve action potentials (SNAPs) and repetitive nerve stimulation (RNS; Figure 3(e)). EMG demonstrated normal insertional activity, recruitment, amplitudes, and interference pattern. The third EMG performed 2 months after onset of symptoms showed some small amplitude motor unit potentials (MUPs), which raised the possibility of a concurrent delayed myopathic process with AMAN.

EMG/NCS and RNS. EMG/NCS were performed at three different time points, that is, at 5D, 14D, and 44D SP onset of symptoms. (a–c) Decreased in the compound of muscle action potentials (CMAP) in the right median and peroneal nerves at three different time points with preserved sensory NCS (not shown). (d) F-wave latency of the median nerve was normal (other nerves tested not shown). See Supplementary Table 2. (e) RNS of the median nerve failed to show a decrement response (other nerves tested not shown).
Moreover, the patient underwent a left quadriceps biopsy (at 35D SP onset of symptoms) to evaluate for myositis and inflammatory nerve changes. The pathology showed an inflammatory process suggestive of small vessel vasculitis, with scattered interstitial small vessels showing transmural inflammation and endothelial destruction with no evidence of thrombosis or fibrinoid necrosis (Figure 4). Many fibers were mildly atrophic with no evidence of fiber type grouping atrophy suggestive of disuse. Trichrome staining showed no myofiber fibrosis, and immunostaining showed perivascular inflammatory cells.

Pathology findings of the left quadriceps biopsy. H&E stains of (a) Cuffing and transmural vascular involvement. (b) Perivascular lymphocytic cuffing. (c) Isolated myonecrosis with no evidence of thrombosis or fibrinoid necrosis. (d) Trichrome stain showing mildly atrophic fibers with no evidence of fiber type groups atrophy suggestive of disuse. Immunohistochemistry stains for (e) CD3 maker for T cells (f) CD4 marker for T helper cells, (g) CD8 marker for cytotoxic T cells, and (h) CD20 marker for B cells.
Re-staging studies demonstrated systemic disease progression, and the patient was started on the BRAF and MEK inhibitors dabrafenib 150 mg per oral (PO) BID and trametinib 2 mg PO daily, respectively (at 21D SP onset of symptoms). At 29D SP onset of symptoms, the patient’s overall strength had improved. Her dyspnea had resolved, and she was able to sit in bed unassisted for 30 min, although she remained quadriplegic and was discharged to an inpatient rehabilitation facility. Several months after the onset of her motor decline, she had resolution of the left facial cranial palsy, and she was able to walk unassisted for 20 to 50 ft. Yet, she never returned to her motor baseline.
Two weeks after discharge from the hospital, she had a symptomatic intracranial hemorrhage in the site of her known MBM in the left temporal lobe (Figure 1). She underwent an emergent craniotomy for evacuation of the hematoma and tumor resection. Given progression of her metastatic disease, she subsequently received radiation therapy (RT) to her lower thoracic spine (2000 cGy in five fractions) and fractioned stereotactic radiation therapy (FSRT, 2500 cGy in five fractions) to the left temporal cavity, followed by whole brain radiation therapy (8/10 treatments) with adjuvant temozolomide several months later (Figure 2). She died approximately 8 months after her MBM diagnosis and 7 months after developing AMAN due to disease progression. An autopsy was declined by her mother.
Discussion
While the syndrome of AMAN is rare, the spectrum of neurological irAEs associated with ICIs is rapidly growing.1,8–23 The induction of these syndromes can help determine the pathophysiology of existing disorders as well as to describe new and rare ones such as those associated with ICIs. Evaluation of these patients can be difficult and a formal neurological consult with appropriate diagnostic studies (i.e. EMG/NCS, CSF, and serum studies) is strongly encouraged.
Non-neurologic Grade 3 and 4 irAEs in melanoma have been well described.1,5,8,11,13,16,18–20,22 However, neurological irAEs are less defined and can be serious and dose limiting. The Checkmate 204 study reported neurological irAEs in 8% of patients, regardless of brain metastases status, 5 many of which were expected in brain metastases (e.g. headache, edema) and could have been exacerbated with study treatment, while others (i.e. hypophysitis 5% and our AMAN case) are likely due to ICIs. Our report of AMAN adds to the list of rare neurologic irAEs reported in the Checkmate 204 study. 5
A wide range of other neurologic syndromes have been previously reported, including a single case of AMAN and few others of acute (aka Guillain–Barré syndrome (GBS)) and chronic inflammatory demyelinating polyradiculoneuropathies (AIDP and CIDP, respectively).10,21,24,25 Most of the neurological irAEs had a median onset of symptoms 1 to 12 weeks after starting treatment,10,21,24 with the subset of GBS patients having onset of symptoms 35 to 84 days after Ipi alone10,21 or 21 to 49 days after Ipi and Nivo.21,24 GBS cases had a wide range of outcomes from full recovery to death following immunosuppression (i.e. steroids, IVIG, or other).10,21,24 The AMAN case was reported after Nivo monotherapy, 23 with onset of symptoms 11 weeks and died 25 days after onset of symptoms. For comparison, our AMAN patient had onset of symptoms 30 days after initiation of Ipi and Nivo, fitting within the window previously noted in these cases, had significant clinical improvement, and died several months later due to progressive systemic metastatic disease. Based on these few reports, it is unclear what the best treatment approach should be for these irAEs. To our knowledge, our patient is the first case with AMAN associated with Ipi and Nivo, who significantly improved after immunosuppressive therapy.
Considered to be an axonal variant of GBS,26–29 AMAN has a similar immune-mediated pathophysiology,29–31 but with the axolemma as the main foci of attack as opposed to the myelin sheath (and related constituents) in GBS.29,30 In both, CSF shows elevated protein with normal WBC count (aka albuminocytologic dissociation) 32 ; however, antibodies to GM1 and GD1a gangliosides are only associated with AMAN, albeit can be negative sometimes.33,34 Since it is challenging to distinguish AMAN from GBS clinically, 35 EMG/NCS is critical for diagnosis, which requires decreased distal CMAP amplitude <80% of the lower limit of normal (LLN) in ⩾2 nerves with no evidence of demyelination, or in a single nerve if distal CMAP < 10% of the LLN.36–38 A rapidly reversible conduction block can be present early in the course of the disease without temporal dispersion, which can be mistaken for a demyelinating pathology.39–42 All GBS variants include treatments with plasmapheresis (PLEX) or IVIG. But patients with AMAN usually have a worse prognosis, partially due to the slow or irreversible process of axonal regeneration. 29
Our patient had all the typical features of AMAN, except for an increased protein with pleocytosis in CSF. This latter feature perhaps relates to both the nature of her metastatic disease causing a breach in her blood brain barrier, and the mechanisms underlying the treatments with ICIs provoking a surmounted response of lymphocytes in the nervous system. Her initial clinical picture could have been misconstrued as a GBS if it were not for the utility of EMG/NCS to arrive at the appropriate diagnosis. It is important to note that neurological irAEs can present with overlapping diagnoses. 43 For this reason in our patient, pyridostigmine was given on initial treatment given concerns for possible overlap with myasthenia gravis. Moreover, findings in muscle biopsy suggestive of small vessel vasculitis allude to an underlying inflammatory process, which may have been similar to a small vessel vasculitis process or myopathy. However, in our patient there was no evidence of myositis, with serum CK levels and EMG studies found normal on repeated occasions, except for the presence of small amplitude MUPs on the third EMG raising the possibility of a parallel myopathic process. The patient had a temporal asymmetric resolution and worsening that might be related to the ICIs, which differentially affect different organs, nerves, and muscles at various times and regions. Limited examination on EMG may not have obtained the typical myopathic changes, which are small, short duration, polyphasic waves with increased recruitment. Her acutely progressive weakness was not explained on findings on MRI of the spine.
It remains unclear what is the pathophysiology behind these rare inflammatory axonal and demyelinating polyradiculoneuropathies that are rising in the setting of ICIs therapy. It is unknown whether their emergence is due to unidentified antigens found in tumor and normal tissue via molecular mimicry or secondary to an autoimmune reaction unleashed by ICIs. Further studies are needed to explore their mechanisms to develop superior treatment strategies. In our patient, given the prompt suspected association of her acute motor decline with ICIs, she was immediately treated according to existing guidelines for immunosuppression (i.e. steroids, IVIG, infliximab), with a significant improvement in motor function several months later. Of note, PLEX has been used in patients with Ipi-induced CIDP, transverse myelitis, and concurrent myositis and MG syndrome, with a reported significant clinical improvement within 2 weeks of starting PLEX therapy. 17 However, PLEX was not given in our case due to concern of decreasing efficacy of the other treatments (i.e. IVIG) and inconclusive data regarding its efficacy in previously reported cases.8,9,12,14,15,17,44,45 Moreover, by preventing the production of pro-inflammatory cytokines (i.e. IL-1, IL-6), anti-TNF-alpha (i.e. infliximab or other FDA-approved biosimilar) is recommended in patients with severe or life-threatening steroid-refractory irAEs from ICI treatment,46–48 without altering the tumor kinetic response or durability, 49 and may confer better outcomes with immunosuppression and/or by avoiding long-term use of corticosteroids,46,50 including neurological irAEs (e.g. audiovestibular dysfunction, transverse myelitis).51–53 The use of infliximab should be considered on an individual basis.
It is important to note that patients with positive BRAF mutated melanoma can benefit from BRAF/MEK inhibitors, which historically have been often used as first line therapy. While targeted agents have a higher response rate, the duration tends to be limited. Since the advent of immunotherapy showed the potential to elicit both a durable response and high response rates, this was the best treatment approach for this young patient with advanced, unresectable metastatic melanoma.54,55
Since the FDA approval of Ipi and Nivo in metastatic melanoma, 1 a growing number of patients are being treated with ICIs. Therefore, the number of these neurologic irAEs is expected to rise, as well as the demand to care for these patients. 56 As the field of immunotherapy in oncology expands, it is critical to increase our knowledge and familiarity with these rare irAEs, and to improve guidelines to optimize their management. Of note, even though this case was part of a large clinical trial study, the delayed adverse effects from immunotherapy in this case were not published in the large clinical trial publication.
A major limitation of our current report is that a cause-effect relationship cannot be established, particularly as the pathophysiology is unknown and there is no reliable marker to prove it was induced by ICI therapy. Nevertheless, this patient allows us to raise awareness about the several neurological irAEs,1,9,10,12,14,15,17,21,23,44 aid with pharmacovigilance, and suggest hypotheses that may help in the development of future clinical trials to predict and manage these irAEs.
Conclusion
The current case is the first case reported who developed AMAN as an irAE associated with combination therapy with Ipi and Nivo, who significantly improved clinically after immunosuppressive treatment. As the field of immune-oncology advances with a rapidly growing use of immunotherapies to treat numerous types of cancers, it is critical to increase awareness about ICI associated irAEs, especially of those less common neurological conditions that are difficult to recognize, diagnose, and treat. These emerging neurological irAE need to be reported to assist in the development of improved guidelines in the management of these devastating, yet potentially reversible complications.
Supplemental Material
sj-pdf-1-sco-10.1177_2050313X211042215 – Supplemental material for Acute motor axonal neuropathy after ipilimumab and nivolumab treatment in melanoma brain metastases: A case report and review of the literature
Supplemental material, sj-pdf-1-sco-10.1177_2050313X211042215 for Acute motor axonal neuropathy after ipilimumab and nivolumab treatment in melanoma brain metastases: A case report and review of the literature by Yolanda Piña, Brittany R. Evernden, Nikhil Khushalani, Kim Margolin, Hussein Tawbi, Nam D. Tran, Robert Macaulay, Peter Forsyth and Edwin Peguero in SAGE Open Medical Case Reports
Footnotes
Authors Note
Nikhil Khushalani is now affiliated to Cutaneous Department. H. Lee MCC and Research Institute. Robert Macaulay is now affiliated to Neuro-Pathology Department H. Lee MCC & Research Institute, Tampa, FL, USA.
Author contributions
All authors of this manuscript contributed to conception and design, data collection, imaging review, manuscript drafting, interpretation of data, conceptual refinement, and critical manuscript review.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval for this study was obtained from the Institutional Review Board (approval number Pro00015053 / ID 18068).
Informed consent
Written informed consent was obtained from the subject before the study. The mother of the patient provided written informed consent to publish this case.
Trial registration
NAME OF TRIAL REGISTRY: TRIAL REGISTRATION NUMBER NCT02320058. Funded by Bristol-Myers Squibb and the National Cancer Institute; CheckMate 204.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.