
Abstract
Heat shock proteins (HSPs) are primarily induced in response to stress stimuli. In cardiovascular diseases, HSPs are expressed pronouncedly and act to protect the cardiac tissue by inhibition of cellular apoptopic mechanisms. An ischemic condition preceded by angina prevents degeneration of tissue by ‘ischemic preconditioning’ in which HSPs produced in an earlier response have a protective effect towards subsequent cardiac injuries. This review highlights the manifestations of HSPs upon stress conditions of the cardiovascular system. The distinctive features of different HSP families in view of their unique behavior in maintenance of stressed cardiac tissues are briefly described. A correlation of the fundamental roles of HSPs in certain cardiac pathological conditions is also discussed. A proteomic approach to study the interactions of HSPs is also documented. Hence, an understanding of the physiological mechanisms of HSPs in cardiac stress offers potential solutions for management of cardiac pathologies and to undertake suitable prophylactic measures.
Keywords
Introduction
Cardiovascular disease is a critical illness affecting a major population with deterioration of cardiac tissues leading to consequent death. Cardiac stress arises from many degenerative heart diseases comprising of acute and chronic myocardial infarction (MI), atherosclerosis, spasm, thrombosis, cardiomyopathy and congestive heart failure. It is evident that excessive stress plays a key role in the pathogenesis of ischemic heart disease. Hence, the prevention of stress-induced myocardial injury is a fundamental process in the prophylaxis of such diseases.
The heart possesses a remarkable ability to adapt itself against any stressful situation by increasing resistance to adverse consequences. This phenomenon, known as ‘ischemic preconditioning’, is an inherent ability of the myocardium to protect itself from recurrent ischemic damage. Ischemic preconditioning is the manifestation of earlier stress response that occurs during repeated episodes of brief ischemia and reperfusion, and can render the myocardium more tolerant to subsequent lethal ischemic injury (Flack et al. 1991). This adaptive protection was first described by Murry et al. (1986) and has been found to be mediated by gene expression and transcriptional regulation (Das et al. 1994; Maulik and Das, 1996). Preconditioning involves a cascade of stress signals such as activation of protein kinase C (PKC), protein tyrosine kinases (PTKs) and mitogen-activated protein kinases (MAPKs). These kinase modulations effect the opening of KATP channels, expression of a number of protective proteins known as heat shock proteins (HSPs), upregulation of nitric oxide synthase (NOS), induction of cyclooxygenase-2 (COX-2) and cellular antioxidants (Baines et al. 2001).
In humans, ischemic preconditioning has been shown to occur in patients undergoing coronary angioplasty with significantly less ST segment deviation, lower mean pulmonary artery pressure, lower cardiac vein flow and less myocardial lactate production during the second balloon inflation (Deutch et al. 1990). Acute MI with antecedent angina patients have a lower mortality, a small infarct size and a lower incidence of complications than patients whose acute MI was not preceded by angina (Kloner et al. 1995). Also, a previously ischemic episode in patients with symptomatic significant coronary artery disease or variant angina appeared to lessen the severity of a subsequent ischemic episode (Tzivoni and Maybaum, 1997) and reduced the frequency of ventricular arrythmias during recurrent vasoplasm (Pasceri et al. 1996). Also, it has been suggested by Bartling et al. (2003) that ischemic preconditioning conserved the expression of cardioprotective determinants in aged human hearts despite limited functional recovery. Recent advances in molecular tools such as gene expression profiling have been employed to study cardiac diseases by differential display, serial analysis of gene expression (SAGE), subtractive hybridization and gene microarrays. Simkhovich et al. (2003) have reviewed this technique for investigation of myocardial ischemia related to biochemical responses, changes in gene expression patterns and activation of genetic survival of the ischemia/reperfused myocardium. In a recent report, Gong et al. (2008) suggest the use of sublethal laser-induced hyperthermia to precondition cardiac tissue against ischemic-induced myocardial infarction. Other stress conditions also affect the cardiac tissues significantly. A recent study by Rodrigo et al. (2008) describes that oxidative stress is a major contributory factor representing the consequences of ischemia/ reperfusion cycle in atrial fibrillation, a common complication of cardiac surgical procedures. It is also reported that in arrythmias, reactive oxygen species (ROS) is involved in its pathogenicity. Overall, this review is aimed to summarize the important roles of heat shock proteins in response to cardiac stress and in certain other cardiac disorders.
‘Classic’ and ‘Delayed’ Preconditioning
The duration of the ischemic stress is an important determinant of the resulting tissue damage, in which short periods of ischemia are well tolerated while prolonged periods induce significant cell death (Loor and Schumacker, 2008). The early form of ischemic preconditioning, referred to as, ‘classic’ preconditioning is observed immediately following brief sublethal ischemia and conferred a marked slowing of the progression of ischemic injury during subsequent ischemia. A distinctive feature of classic preconditioning is that the protection afforded by antecedent ischemia rapidly lapses out. In most subjective models, no protection against infarction was observed if the preconditioning and the subsequent ischemic insult extended beyond 2 h period and is therefore, shortlived.
A delayed form of adaptation is manifested subacutely around 24 h following preconditioning with ischemia, variably known as, ‘delayed’ preconditioning; ‘late’ preconditioning; ‘second window’ preconditioning or ‘second window of protection’ and is observed as a protection against a variety of ischemic pathologies (Baxter and Ferdinandy, 2001).
Cardiovascular Chaperones
Molecular chaperones are essential components of the cardiomyocytes, constitutively expressed in minimal levels for normal cardiac function and are raised in terms of inducible expression in response to cardiac stress. Molecular chaperone activity is the primary function of HSPs. HSPs are structurally and functionally well-conserved stress proteins among species and are triggered by a variety of cellular stress stimuli resulting in their enhanced synthesis, while under normal physiological conditions, they are present only in basal amounts. HSPs are generally upregulated in heart in response to Ischemia/Reperfusion (I/R), hemodynamic overload and congestive heart failure and mediate cardioprotection through biological functions as molecular chaperones (Agashe and Hartl, 2000). HSPs also display antioxidant effects, anti-inflammatory actions, aid in folding and refolding of proteins, protect membranes from damage and inhibit apoptosis (Hooper and Hooper, 2005). More recently, HSPs are also known to act as cell surface receptors (Calderwood et al. 2007). The predominant roles of HSPs are described in Table 1. The consensus appears to be that loss-of-function of HSPs compromises the ability of the heart to handle stress, while HSP gain-of-function is cardioprotective (Kumarapeli and Wang, 2004). Intracellular and membrane-bound HSPs have been identified, either offering prevention to lethal damage or act as danger signals and elicit immune response (Multhoff, 2007). Recently, identification of cell status has been determined as an effective criterion for measuring the secretion of HSPs by Ireland et al. (2007). This also implies that the analysis and identification of ligands secreted with HSP can be quantified by the protocol. Another report by Vigh et al. (2007) indicates that membrane microheterogeneity is important for regulation of HSP response. HSPs are implicated in various cardiac pathological conditions. To note some; ischemic preconditioning, myocardial hypertrophy and cardiomyopathy tend to upregulate an array of HSPs for eventual cardioprotection. This article therefore intends to provide an insight into these consequences and briefly describe the potential roles of HSP response. We describe here the major HSP families of 110 kDa, 90 kDa, 70 kDa, 60 kDa and low molecular weight stress proteins. HSPs are conventionally represented according to their molecular mass. The HSP response to various cardiac stress is represented in Figure 1.
Protective roles of heat shock proteins.

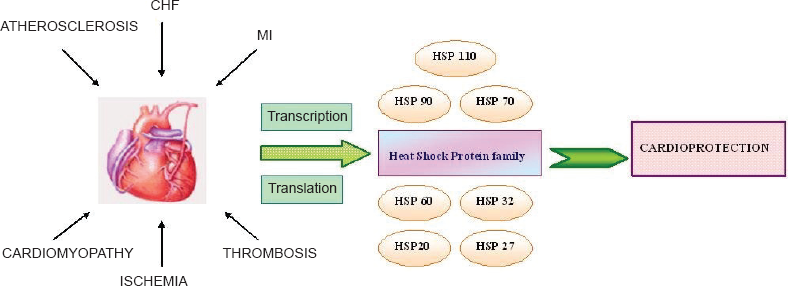
Activation of HSPs in cardiac stress. Stress signals to the heart elicit transcription and subsequent translation of HSPs to effect cardioprotection.
HSP110 class
The HSP110 family comprises of constitutively expressed HSP110 and GRP78, ORP150 inducible specifically by hypoxia, and, HSP94 inducible by heat and osmotic stresses (Snoeckx et al. 2001). The amino acid composition of the mammalian ER HSP110 family shows weak correlation with GC3 content. Pro (P), Ala (A), Arg (R), and Gly (G) distributes approximately 4%-9% of codon usage, while Phe (F), Ile (I), Met (M), Tyr (Y), Asn (N), and Lys (K) distributes approximately 2%-10%, in the amino acid sequences of GRP78 and ORP150 of the HSP110 family (Takeuchi and Mala, unpublished).
HSP70 class
HSP70 is the most abundant and best-characterized heat shock protein, first observed with hyperthermia (Mizushima et al. 2000). In the mammalian myocardium, elevation of HSP70 upon heat stress and ischemia results in development of tolerance to subsequent severe ischemic stress (Yellon and Latchman, 1992; Marber et al. 1993). Hypoxia, oxygen radicals and proinflammatory cytokines stresses also induce HSP70 secretion (Moseley, 1997; De Maio, 1999). It has been observed that pre-induction of HSP70 by heat stress had various other beneficial effects after I/R injury (Saad et al. 1995).
Constitutive and inducible HSP70s are involved in cardiac preservation during oxidative stress (Su et al. 1998). It has been hypothesized that HSP70 may protect cells by processing denatured proteins and by protecting RNA processing and subsequent translation (Jaattela and Wissing, 1992). HSP70 is involved in antigen processing and presentation, in acute phase response and cytokine production (Minowada and Welch, 1995) and also inhibits the production of proinflammatory cytokines (Simon et al. 1995). Further, HSP70 is related to disease severity with a protective role, but, however, does not predict survival (Genth-Zotz et al. 2004). All members of the HSP70 family exhibit a common structure consisting of two domains; a highly conserved amino-terminal ATPase and a carboxy-terminal peptide-binding domain (Minowada and Welch, 1995).
HSP70 is also a powerful mediator of inflanimation and immunity. However, conventional mechanisms do not aid HSP70 in traversing the plasma membrane, as it lacks a consensus secretory signal. Another mechanism of HSP70, independent of
HSP72, an inducible form of the HSP70 family is strongly induced in the myocardium under various stress conditions of hyperthermia (Currie and White, 1983), hypoxia (Howard and Georghegan, 1986), acute cardiac overload (Delcayre et al. 1988) and ischemia (Yellon and Latchman, 1992). Induction of HSP72 confers resistance to these stress conditions and plays a pivotal role in protection of cells from subsequent myocardial injury. Comini et al. (1996) have also demonstrated the induction of HSP72 upon congestive heart failure. HSP72 is referred as a ‘chaperokine’ to describe its unique function as a chaperone as well as a cytokine. Intracellular HSP72 is generally cytoprotective by induction of the cells' anti-apoptotic mechanisms, repression of gene expression, modulation of cell cycle progression and anti-inflammation. Extracellular HSP72 is also immuno-stimulatory, stimulates pro-inflammatory cytokine synthesis, augments chemokine synthesis and enhances anti-tumor surveillance. HSP72 is released in a free form and within highly potent exosomes. HSP72 has a profound effect on host immunity and its circulation primes the immune system to real or perceived danger (Asea, 2007).
HSP90 class
HSP90 is one of the most abundant constitutively expressed stress protein and is located in the nucleus and cytoplasm of eukaryotic cells. HSP90 is also heat-inducible (Ketis et al. 1993) and more strongly inducible by I/R injury (Nishizawa et al. 1996). Generally, HSP90 is well-conserved among prokaryotes and in eukaryotes (Pearl and Prodromou, 2000; Pearl and Prodromou, 2006). HSP90 is known to activate production of agonist-stimulated release of NO and subsequent endothelium-dependent relaxation of blood vessels (Garcia-Cardena et al. 1998). In addition, HSP90 associates with transcription factors which modulate transcriptional activity and expression of other heat shock proteins (Nadeau et al. 1993). Further, HSP90 aids in the modulation of cytoskeletal dynamics of the stressed cells to attain myocardial protection (Gray et al. 1999). HSP90 contains a catalytic loop that accepts the y-phosphate of ATP in its core domain and is therefore characterized to be a split ATPase (Meyer et al. 2003). However, HSP90 is not capable of independent functionality as a protein chaperone, but requires the augmentation of its co-chaperones. HSP90 is crucial in signal transduction to transformation to genetic capacitance and influences a wide array of cellular events (Brown et al. 2007).
HSP60 class
HSP60 is constitutively expressed in the cytoplasm and translocated to the mitochondria (Gething and Sambrook, 1992). HSP60 is also heat inducible, although, ischemia has been shown to be a potent inducer (Marber et al. 1993). This class of stress proteins is a major molecular chaperone of the mitochondria. HSP60 facilitates the refolding of mitochondrial proteins as they translocate the inner and outer mitochondrial membranes (Ostermann et al. 1989). HSP60 consists of seven subunits with a weak K+-dependent ATPase activity (Gray et al. 1999). By constitutive expression, HSP60 also plays an essential role in normal cell function (Latif et al. 1999). In atherosclerotic models, autoimmunity to HSP60 indicated a plausible role in aetiology of the disease (Hoymans et al. 2007).
Small heat shock protein (sHSP) family
The small HSP subfamily also represents one of the best studied stress proteins (Kumarapeli and Wang, 2004). In earlier studies, Lam et al. (1996) have isolated and characterized cDNA of human heart encoding HSPL27, a novel member of sHSPs, named 27 kDa heat shock protein-like protein. Another new member of the sHSP family was identified as HSPB2, with a genomic locus less than 1 kb from the 5'-end of the ≈B-crystallin gene with opposite transcription direction. It is also reported that HSPB2 gene was expressed preferentially in skeletal muscle and heart (Iwaki et al. 1997).
In mammalian species, sHSPs comprise of: HSP27, HSPB1, HSPB2, HSPB3, ≈A-crystallin (HSPB4), ÃB-crystallin (HSPB5), HSP25, HSP20 (HSPB6), HSPB7, HSP22 (HSPB8), HSPB9 and HSPB10 (Kappe et al. 2003). The sHSPs have been classified into two main categories: Classes I and II, according to their different patterns of gene expression and subcellular localization (Taylor and Benjamin, 2005). All sHSPs share a common ≈-crystallin domain with unique N-terminal and C-terminal extensions and the latter is critical for their chaperone activity (Pasta et al. 2002).
HSP27 is fundamental to ischemia-induced delayed myocardial protection (Baxter and Ferdinandy, 2001; Efthymiou et al. 2004). I/R injury also induces overexpression of HSP27 which acts to substantiate cardioprotection after repetitive insults (Wang et al. 2007). Earlier studies have well-demonstrated the cardioprotective roles of HSP27 in isolated hearts
HSP20 is a molecular chaperone which functions to assist proteins in achieving and maintaining proper conformation (Van de Klundert et al. 1999). HSP20 enhances cardiac function and renders cardiac protection against β-agonist-mediated apoptosis and I/R injury (Fan et al. 2005). HSP20 has also been linked to calcium handling. Cardiomyocyte permeabilization with HSP20 results in a significant increase in cell shortening and a decrease in calcium transient values (Pipkin et al. 2003). Additionally, HSP20 increases the amplitudes of both calcium transients and cell contraction in cardiomyocytes (Chu et al. 2004). Recently, Islamovic et al. (2007) have investigated the C-terminal extension of HSP20 to provide a mechanistic insight into its possible function. I/R injury resulted in cardiopro-tection by HSP20 only by overexpression of full-length HSP20, while substitution of its C-terminal did not show any protectivity.
HSP32 [Heme oxygenase -1 (HO-1)] is a unique protein (acting on heme) and is induced in response to oxidative stress. HSP32 acts as a mediator of cytoprotection in ischemic heart disease (Ozono, 2006), delayed myocardial preconditioning (Jancso et al. 2007) and cardiovascular damage by inhibition of oxidative stress and offers cellular protection. Phosphorylation by activation of protein kinases and hypoxia has also been reported to induce HSP32 in cardiomyocytes (Wu et al. 2004). HSP32 plays a cytoprotective role and exerts antiinflammatory, antiapoptotic, antioxidant effects, and is also recently known to possess proangiogenic properties (Dulak et al. 2008). In a recent study, HSP32 has been reported to inhibit postmyocardial infarct remodeling and influence the restoration of ventricular function (Liu et al. 2006).
Gene Expression and Regulation of Heat Shock Proteins
Activation of the heat shock gene is rapid in response to heat stress or physiological stimuli. In the stressed cell, heat shock transcription factor (HSF) becomes phosphorylated and assembles to form activated trimers in the cytoplasm, accumulates in the nucleus, and associates with specific target sequences on the DNA promoter region of the heat-inducible gene, known as the heat shock element (HSE). The binding of the HSP trimer to the HSE results in transcription of HSP mRNA. Transcription of heat stress protein gene is also modulated by other transcription factors, such as, TATA-binding protein and the GAGA factor (Fig. 2).

Schematic diagram of gene expression of HSPs. Upon stress, heat shock factors (HSF) accumulate in the cytoplasm and form active trimers by phosphorylation of Ser and Thr. HSF trimers traverse into the nucleus and bind to the heat shock element (HSE), a regulatory sequence upstream of the HSP gene, and activate RNA Pol II for synthesis of the mRNA transcript. The activation of RNA Pol II may also be effected by activation of GABA and TATA sequences by transcription factors GABA factor (GABAF) and TATA-binding protein (TBP). mRNA transcripts reach the cytoplasm where normal translation takes place in the ribosome unit (enlarged) with tRNAs.
The next level of regulation occurs at translation of the heat shock protein mRNA (Fig. 2). Heat shock genes do not contain introns or the intervening sequences, a unique feature of eukaryotic genes, which allow rapid transcription of the gene and mRNA processing. As a result, the response time for HSP expression is quick and efficient. Upon heat stress, HSP mRNA accumulates in the cytoplasm, ribosomes bind to the template and HSPs are the products of protein synthesis (Gray et al. 1999).
Differential Expression of Heat Shock Proteins
It has been observed by Knowlton et al. (1998) that disparate regulation of HSP levels occurred in failing hearts in comparison with the normal myocardium. Both HSP27 and HSP60, associated with the cytoplasm and mitochondria, elevated in dilated cardiomyopathic ventricles, while only HSP60 was similarly elevated in ischemic cardio-myopathic ventricles. In contrast, no significant increase of HSP72 or HSP90 was known to occur in both conditions of the failing hearts. Thus, a greater correlation of different HSP levels prevailed in both cardiomyopathies than in normal hearts.
Stress-Induced Cardioprotective Effects of Heat Shock Proteins
It was observed by Dillman et al. (1986) that hearts subjected to prolonged ischemia and subsequent necrosis, expressed increased levels of inducible HSP70. Currie et al. (1988) introduced the concept of heat stress preconditioning as a strategy for myocardial infarction. It was therefore proposed that heat stress preconditioning was beneficial for the ischemic myocardium (Gray et al. 1999). Subsequently, Mizushima et al. (2000) reported that preinduction of HSP70 protected cardiovascular function following trauma-hemorrhage and resustication. Contemporary reviews by Latchman (2001) and Snoeckx et al. (2001) provide a wealth of information on the protective roles of HSPs in myocardial stress.
Between 1995-1996, it was demonstrated that overexpression of HSP70 protected the heart against the damaging effects of ischemia using a variety of determinants such as infarct size, creatine kinase release, recovery of high energy phosphate stores and correction of metabolic acidosis (Marber et al. 1995; Plumier et al. 1995; Radford et al. 1996). The cardioprotective mechanisms of HSPs involve the inhibition of mitochondrial caspase 9 pathway of apoptosis by HSP27 (Bruey et al. 2000), HSP90 (Pandey et al. 2000) and HSP70 (Beere et al. 2000; Saleh et al. 2000) as described by Latchman, (2001). HSP70 also acts by inhibition of c-Jun N-terminal kinase (JNK) in a caspase independent apoptosis (Gabai et al. 2000). HSP27 can protect the integrity of the microtubules and actin cytoskeleton in cardiac myocytes (Bluhm et al. 1998) and endothelial cells (Loktionova et al. 1998) exposed to ischemia. Also, HSP90 has been shown to bind to endothelial nitric oxide synthase and stimulate its activity (Garcia-Cardena et al. 1998). Further, the overexpression of HSP70 enhanced NO production in response to cytokine stimulation (Bellmann et al. 2000). Cardioprotective effects of heat shock proteins during cardiac surgery in pediatric patients have also been described by Giannessi et al. (2003). HSP70 has also been identified as a cytoprotective therapeutic agent (Rokutan et al. 1998) which decreases the risk of postoperative atrial fibrillation by reducing I/R injury (Rammos et al. 2002). Mild-to-moderate alcohol consumption is also associated with an induction of the expression of several cardioprotective proteins such as heat shock and antioxidant enzymes. Thus, the upregulation of these proteins may explain the cardioprotective ability of alcohol (Sato et al. 2004). Of recent interest, Kwon et al. (2007) have analyzed the efficiency of protein transduction domain (PTD)-mediated delivery of HSP27 on I/R injury. This represents a potential therapeutic strategy as protein drug for ischemic heart diseases.
Free radicals formed by oxidative stress in myocardial I/R injury, result in ventricular dysfunction, or “myocardial stunning,” arrhythmias, and progressive cell damage or death after ischemic injury. Molecular chaperones also effect cardioprotection to counteract the reactive oxygen species (ROS) by mediating the protective roles of glutathione peroxidase (GPx), superoxide dismutase (SOD) and catalase (Benjamin and McMillan, 1998).
Cell culture models have also been very helpful in unraveling the expression patterns and mechanisms of cardiac protection of HSPs. The existence of protection in cultured adult or neonatal cardiomyocytes against a severe, lethal stress challenge after previous activation of HSP-gene transcription has been described (Snoeckx et al. 2001).
Cardioprotection of Heat Shock Proteins in Vascular Endothelial Cells
Heat shock proteins have an extremely significant function in the maintenance of hemodynamic stress of the vascular cells (Xu and Wick, 1996). Coronary endothelial cells are the main site of induction of 70 kDa heat shock protein in the heart and appear to contribute to the protective effects of heat stress on the recovery of mechanical and endothelial function (Amrani et al. 1998). The consequences of cardiac ischemia—reperfusion are not limited to myocytes but also extend to the coronary endothelium (Laude et al. 2002). Thereby, similar to cardiomyo-cytes, the endothelial cells also express heat shock proteins in response to cardiac insults. It has also been observed that, in vascular lesions, an immune response is elicited towards heat shock proteins and elevated levels of anti-hsp antibodies could trigger ischemic stroke (Gromadzka et al. 2001).
Heat Shock Protein Pathways in Cardiac Stress
Heat shock proteins are mostly inducible and several pathways co-exist for the induction of HSPs. Identification of signaling pathways for induction of HSPs rely on various cellular components and is still a poorly understood phenomenon. Kacimi et al. (2000) have investigated the role of kinases in hypoxia-induced HSP expression and their plausible effects on HSP induction. The kinases that contribute to the induction of HSPs include protein kinase C and the mitogen-activated protein kinase family (MAPK), that comprise of extracellular-signal regulated kinase (ERK), c-Jun NH2-terminal kinase and the stress-activated protein kinase (JNK/SAPK) and p38. Hypoxia leads to activation of multiple kinase cascades in the cardiomyocytes (Kacimi et al. 1998). p38 stress kinase appears to play a direct role in the regulation of HSP32 expression. In contrast, HSP70 is strongly affected by the multiple kinase cascade signaling. Hypoxia induced phosphorylation of MAPKAP-2/3 activated kinases and HSP27 by the activation of signaling pathways downstream of p38 stress kinase. Thereby a differential pattern of regulation of HSPs has been observed by the kinase cascade signaling mechanisms.
ROCK (Rho-associated kinase) pathway is implicated in cardiovascular aberrations and is detrimental to the heart (Noma et al. 2006). However, this pathway has no direct role on HSPs, but inhibition of this pathway activates the endothelial nitric oxide synthase (NOS) which has a protective effect on the vasculature.
Any defect in the pathways of induction of HSPs may lead to deprivation of the protective mechanisms following cardiac stress and eventually lead to apoptosis and increase the potency of cardiovascular risks. Therefore, strategies requiring the modulation of HSP pathways are still to be investigated, also for exploitation of therapeutic measures in cardiac stress.
Heat Shock Proteins in Patho-Physiologies of Cardiac Stress
Cardiac stress also results from various pathophysiological factors eliciting a number of stress proteins. Snoeckx et al. (2001) have described important relations of these pathological insults of the cardiac tissues and the strategic responses of HSPs. An important factor in considering the potential of HSPs to improve cardiac function during and after ischemia is the age of the tissues under investigation. Tissue aging seems to increase susceptibility to cardiac ischemia in aged humans. It is known that overall gene transcription, mRNA translation, and protein degradation were decreased during senescence, whereas a number of malfunctioning proteins were increased (Lakatta, 1993). Because HSP-mediated stress protection could be of special interest in aged individuals, several studies have been initiated to evaluate their potential in this population group. The stress-induced synthesis of HSPs was reduced in the aging cardiovascular system and cardiac HSP70 synthesis upon heat shock was substantially lower than in young rats (Bongrazio et al. 1994). It may be concluded that the heat shock response is reduced in aged hearts and limited HSP synthesis showed reduced protective effects on ischemia tolerance.
Pathological myocardial hypertrophy is acknowledged to be a major risk factor for, myocardial infarction and heart failure (Levy et al. 1990). The pathologically hypertrophied heart is more susceptible to ischemic damage. Sustained poor cardiac output, permanent arrhythmias, and increased loss of intracellular enzymes are commonly observed during postischemic reperfusion of hypertrophied hearts. This poor function is often associated with myocardial contracture and a substantial underperfusion of subendocardial layers of the left ventricular wall (Snoeckx et al. 1990). Understanding the potential of hypertrophied cardiac tissue to upregulate HSP synthesis would be highly valuable, since these proteins could attenuate the poor ischemia tolerance in this type of heart. HSP70 and HSP60 were transiently overexpressed, while low levels of HSP90 were observed (Snoeckx et al. 2001).
Induction of HSPs in tissues to be transplanted is still under investigations, although available reports are scarce. Heat pretreatment of a long-lasting hypothermic storage of cardiac tissue resulted in a significantly improved and accelerated recovery of developed pressure and coronary flow, while the residual ATP and total energy-rich phosphate tissue content was significantly higher than in non-retreated control hearts. HSP70 tissue content has been used as a marker for the risk of rejection of tissue transplants. In transplanted tissues, high HSP levels reflect a response to inflammation, apoptosis, and/or necrosis.
Proteomics of Heat Shock Proteins in Cardiac Stress
The development of proteomics since 1995 (Wasinger et al. 1995), has prompted researchers to investigate its potential application in cardiovascular research. There has been a striking surge of interest for proteomic analysis in cardiovascular biology in view of its current thrust (Arrel et al. 2001; Jiang et al. 2001; Macri and Rapundalo, 2001; McGregor and Dunn, 2003; Borozdenkova et al. 2004; Dohke et al. 2006; Doll et al. 2007). Changes to the cardiovascular system arise from or have the potential to alter, proteomes of cardiac muscle and components of the vascular system, including smooth muscle and endothelial cells. Such changes may be documented through an integrated series of proteomic approaches. A detailed 2-DE analysis of dilated cardiomyopathy—diseased human myocardial tissue revealed more than fifty HSP27 protein species by immunoblotting (Scheler et al. 1997). Protein changes of HSP72, HSP70i, mitochondrial HSP70 precursor, mitochondrial stress protein (HSP70-related), HSP60, mitochondrial matrix protein p1 (membrane-bound HSP60), ≈B-crystallin and HSP27 have been documented by proteomic analysis in cardiovascular diseases (Arrel et al. 2001). The causes of cardiac dysfunction likely result from alternations in cardiac protein expression. Therefore, determining the posttranslational modifications of cardiac proteins in response to cardiomyopathies is important to provide unique insights and an understanding of the mechanisms of cardiac malfunctions. Dohke et al. (2006) have performed the proteomic analysis of cardiac sHSP expression in congestive heart failure (CHF). It was observed that the cardiac sHSPs were highly expressed after the induction of CHF. The increased expression of ≈B-crystallin and HSP27 decreased serum markers of cardiac damage against hypoxic myocardial injury (Martin et al. 1997). HSP20 was significantly increased in CHF compared to the normal heart. It is therefore evident that three cardiac sHSPs, namely, HSP20, HSP27 and ≈B-crystallin play a critical compensatory role in the pathophysiology of CHF (Dohke et al. 2006).
Conclusions
Heat shock proteins could be defined as the ‘guardian’ components secreted in response to various cardiovascular stresses. They have the inherent abilities to protect cardiac tissues from recurring injuries. Constitutive HSPs help in regulation of cells under normal conditions. However, a majority of HSPs are inducible by stress stimuli and play significant roles in preventing cellular apoptosis. The inducible HSPs act as the first line of defense by promoting resistance of tissues upon initial stress and further help to withstand subsequent cardiac malfunctions, by ‘classic’ or ‘delayed’ preconditioning. Most HSPs act as molecular chaperones of the cardiovascular system. Further, the protective potentials of HSPs could be essential to salvage cardiac tissues during repetitive cardiac stress. Therefore, an understanding of the physiological functions of HSPs would be advantageous for development of synthetic drugs for cardiovascular prophylaxis.
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgements
The sponsorship of J. Geraldine Sandana Mala by Kikuji Takeuchi and Naomi Takeuchi of TAK-ENEN, Japan, is gratefully acknowledged.