
Abstract
Circumstantial evidence suggests that oxidative stress plays a crucial role in the initiation and progression of atherosclerosis, but little is known about the relationship between oxidative stress per se, cholesterol transport and endothelial cell integrity. The aim of the present work is to tackle this issue by treating human umbilical vein endothelial cells (HUVEC) with iron/ascorbate for 4 and 8 hours and by subsequently evaluating the cholesterol flux, the gene expression status of cholesterol transporters, nuclear receptors and adhesion molecules, as well as the cellular adhesion of THP-1 monocytes to HUVEC. The incubation of HUVEC with iron/ascorbate resulted in marked lipid peroxidation as reflected by high malondialdehyde levels, which were reduced by pre-treatment with the antioxidant Trolox. Our experiments could not reveal any modifications in the protein and gene expression of the transporters (ABCA1, SR-BI, LOX-1), the adhesion molecules (VCAM-1, ICAM-1 and E-selectin) and the nuclear receptors (PPARs and LXRs) under the influence of iron/ascorbate. However, oxidative stress enhanced monocyte adhesion to HUVEC, induced the gene expression of ICAM-1, E-selectin and MCP-1, whereas it downregulated eNOS mRNA in the presence of monocytes. Overall, our data suggest that oxidative stress is more harmful in the presence of heterocellular communication between endothelial cells and monocytes.
Introduction
Endothelial cells are crucial for the full integrity of the vessel wall. Marked alterations of their function predispose the vessel wall to vasoconstriction, leukocyte adherence, platelet activation, thrombosis, vascular inflammation and the pathogenesis of atherosclerosis. Various studies have emphasized the central involvement of low-density lipoprotein (LDL) oxidation in the endothelial dysfunction, which culminates in the formation of the atherome (Murohara et al. 1994; Libby et al. 2002). In fact, the incursion of oxidized LDL (oxLDL) in significant amounts in endothelial cells induces accelerated proinflammatory effects, including the initiation of the adhesion of leukocytes to the dysfunctional and injured endothelium, and the penetration of these cells into the vessel wall to generate foam cells (Frostegard et al. 1991; Laursen et al. 2001; Hansson, 2001). Mechanistic studies show that transcriptional activation of adhesion molecules, including P-selectin, E-selectin, vascular cell adhesion molecule-1 (VCAM-1), monocyte chemoattractant protein 1 (MCP-1), intercellular adhesion molecule-1 (ICAM-1), and platelet endothelial cell adhesion molecule (PECAM) by nuclear factor-kappa B (NF-κB) plays a pivotal role in the process of cell-cell adhesion (Collins et al. 1995; Pan et al. 1995; Botella et al. 2000; Takehana et al. 2002; Kaur et al. 2003).
If the remarkable capacity of oxLDL to contribute to vascular lesions has been the subject of intensive investigation, little attention has been devoted to the direct influence of oxidative stress per se on the endothelium integrity and metabolic behaviour. However, the increase of reactive oxygen species to high levels may react with cellular constituents to cause severe damage, disruption of function or degradation, as is the case for DNA, proteins and lipid structure (Dargel 1992; Ames et al. 1993). Recently, we explored whether iron-catalyzed free radical-mediated lipid peroxidation provoked abnormalities in cholesterol trafficking in macrophages with an attention on the potential mechanisms (Marcil et al. 2006). We were able to show that exposure of THP-1 derived macrophages to iron/ascorbate induced lipid peroxidation as assessed by the rise of malondialdehyde (MDA), decreased cholesterol efflux, diminished the gene and protein expression of ATP-binding cassette transporter A-1 (ABCA1) among other unaffected receptors that regulate cholesterol homeostasis investigated, and down-regulated the expression of peroxisomal proliferator-activated receptor (PPAR)≈, PPARγ, liver X receptor (LXR)≈ and LXRβ. How endothelial cells respond to oxidative stress, as the unique causal factor, has poorly been examined. Does oxidative stress affect key cholesterol transporters such as SR-BI and ABCA1? Is it able to modify the transcription factors that trigger cholesterol uptake and exo-cytosis? What are the consequences of oxidative stress on the co-culture of monocytes and endothelial cells? Experiments were performed to answer these important and intriguing questions. Our hypothesis was that iron/ascorbate-induced lipid peroxidation may trigger significant disturbances in cholesterol flux and chemotaxis mediators when endothelial vascular cells communicate with monocytes.
Aims
The aims of our study were to explore if oxidative stress affects key cholesterol transporters, regulates transcription factors that trigger cholesterol transport, and modifies the interactions between monocytes and endothelial cells. Our hypothesis was that iron/ascorbate-induced lipid peroxidation may trigger significant disturbances in cholesterol flux and chemotaxis mediators when endothelial vascular cells communicate with monocytes. More precisely, the specific objectives of the present study were to determine the role of iron/ascorbate-mediated lipid peroxidation in cholesterol influx and efflux in endothelial cells, gene and protein expression of scavenger receptor class B type I (SR-BI), ABCA1 and lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), mRNA status of PPAR≈, PPARβ/δ and PPARγ, as well as LXR≈ and LXRβ, and finally the consequences on the interactions of vascular and inflammatory cells.
Methods
Cell Culture
HUVEC (Clonetics™ Human umbilical vascular endothelial cells, Cambrex) were cultured in EGM™ Bullet kit (CC-3124) containing 500 ml Endothelial Cell Basal Medium supplemented with 2 ml Bovine Brain extract, 0.5 ml human epidermal growth factor (hEGF), 0.5 ml hydrocortisone, 10 ml fetal calf serum and 0.5 ml GA-1000 (Gentamicin, Amphotericin). The cell line was cultivated at 37 °C, 95% humidity and 5% CO2 and used between passages 4 and 12 (Takei et al. 2001).
THP-1 human monocytes (American Type Culture Collection (ATCC) TIB 202) were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin and 0.05 mM 2-mercaptoethanol. The cell line was cultivated at 37 °C, 95% humidity and 5% CO2 and used between passages 4 and 12. Cells were differentiated into macrophages by the addition of 100 ng/ml phorbol 12-mysistate 13-acetate (Sigma) for a 72-hour period (Marcil et al. 2006).
Estimation of Lipid Peroxidation
HUVEC cells were incubated in the presence or absence of 100 μM of Fe2+ and 1000 μM of ascorbate and the antioxidants Trolox (6-Hydroxy-2,5,7,9-tetramethyl-chroman-2-carboxylic acid, Sigma) (0.5 mM) and butylated hydroxytoluene (BHT) (0.5 mM) for 4 hours at 37 °C. The reaction was terminated by the addition of 0.2% BHT to measure MDA, as an index of lipid peroxidation. The amount of free MDA formed during the reaction was determined by HPLC as described previously (Courtois et al. 2000). Briefly, proteins were first precipitated with a 10% sodium Tung state (Na2WO4) (Aldrich Chemical) solution, and protein-free supernatant was then reacted with an isovolume of 0.5% thiobarbituric acid (Sigma) solution at 90 °C for 60 min. After cooling to room temperature, chromogene was extracted with 1-butanol and dried over a stream of nitrogen at 37 °C. The dry extract was then resuspended in a KH2PO4/methanol (70:30, pH 7.0) mobile phase before MDA detection by HPLC.
MTT Assay
After the treatment of HUVEC with iron/ascorbate, Trolox (0.5 mM), BHT (0.5 mM) and its carriers dimethyl sulfoxide (DMSO, Sigma) and ethanol (ETOH), the medium was aspirated from the cells and 200 μl of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (0.5 mg MTT/ml PBS) was added to each well. Cells were incubated for 2 h at 37 °C, 95% humidity and 5% CO2 to allow MTT oxidation by mitochondrial dehydrogenase in the viable cells. After 2 h the MTT solution was aspirated and DMSO (200 μl) was added to dissolve the resulting blue Formosan crystals. The absorbance was measured at 535 nm. DMSO was used as blank (Otto et al. 2007).
Isolation and modification of lipoproteins
Human LDL (1.019 < d < 1.063 g/ml), high-density lipoprotein (HDL)3 fraction (1.125 < d < 1.210 g/ml) and lipoprotein-deficient serum (LPDS, d > 1.125 g/ml) were prepared from plasma of healthy human subjects and isolated by differential ultracentrifugation as described previously (Levy et al. 1990a; Levy et al. 1990b; Levy et al. 2000). The lipoprotein fractions were dialyzed intensively against phosphate buffered saline (PBS, pH 7.4) containing 150 mM NaCl and 0.3 mM ethylene diaminetetraacetic acid (EDTA). In order to generate oxLDL, plasma LDL (3 mg apo B/ml) was extensively dialyzed against PBS (pH 7.4) containing 150 mM NaCl and 5 μM EDTA and then incubated with 10 μM CuSO4 for 18 h at 37 °C. Its modification was verified by its mobility on agarose gel electrophoresis (Paragon, Beckman Instruments). All lipoprotein fractions were filtered through a 0.2 μM Millipore membrane and stored at 4 °C.
Cholesterol oxLDL uptake
HUVEC cells were cultured in 6-well plates at 0,5 × 106 cells per well. After a 48-hour period for reaching confluence, cells were exposed for 4 hours to iron/ascorbate with or without the antioxidant substrates and then were incubated for 2 h at 37 °C in 0.5 ml of supplemented EBM with 5% v/v LPDS containing [3H]-cholesteryl hexadecyl ether-oxLDL (12,5 μg/ml). To determine the non-specific binding, cells were incubated with labeled-oxLDL in the presence of a 50-fold excess of unlabeled oxLDL. The assays were essentially carried out as previously described (Suc et al. 2003; Marcil et al. 2006).
Cholesterol Efflux
HUVEC cells and THP-1 macrophages were loaded with radiolabeled cholesterol by incubation for 24 hours in 0.5 ml of supplemented BME with 5% v/v LPDS and 2.64 × 106 dpm/ml [3H]-cholesteryl oleate albumin. After a 16-hour equilibration period of time without radioactivity, cells were washed with PBS and treated for 4 hours with or without iron/ascorbate and antioxidants. Cells were washed again and incubated with HDL3 (50 μg/ml) for 24 hours. The media were centrifuged at 4000 g for 10 min to remove any suspended or dead cells. The cells were washed twice with PBS, resuspended in 1 ml of lysis buffer and homogenized by sonication on ice. The assays were performed as previously described (Suc et al. 2003; Marcil et al. 2006).
Western Blot Analysis
To determine the protein expression of SR-BI, ABCA1, and LOX-1, cells were homogenized and proteins (30 μg) were denatured at 95 °C for 5 min in SDS dithiothreitol and β-mercaptoethanol-containing sample buffer, separated on a 4%-7.5% gradient SDS-PAGE, and electroblotted onto Hybond-C Extra nitrocellulose membranes (Amersham) in 25 mM Tris and 192 mM glycine. Membranes were blocked in Tris-buffered saline [20 mM Tris-HCl (pH 7.5) plus 137 mM sodium chloride (NaCl)] containing 0.1% Tween 20 and 5% non-fat dry milk for 60 min at room temperature. The blots were then incubated overnight at 4 °C in blocking solution with the antibodies for SR-BI (Novus Biologicals) (1:1000), ABCA1 (Novus Biologicals) (1:1000), LOX-1 (Santa Cruz Biotechnology) (1:2000), and β-actin (Sigma-Aldrich) (1:5000). The relative amount of primary antibody was detected with species-specific horseradish peroxidase-conjugated secondary antibody. Blots were developed and the protein mass was quantified using a MultiImage™ Light Cabinet (Alpha Innotech Corporation, San Leandro, CA) and a Alphamager 1220 software (Alpha Innotech Corporation, San Leandro, CA). Bands were analyzed using Scion Image software.
RT-PCR expression analysis
Levels of specific mRNAs were assessed by the reverse transcription-polymerase chain reaction (RT-PCR) under quantitative conditions. Total cellular RNA from HUVEC and THP-1 was isolated using Trizol® Reagent (Invitrogen) according to the manufacturer's protocol. The quantity and yield of the RNA were assessed by the 260:280-nm optical density ratio and by electrophoresis in 1.5% agarose gels and were viewed via ethidium brominde staining. Complementary DNA was synthesized in a total volume of 20 μl from RNA samples by mixing 2 μg of total RNA, 2 μl of reverse transcriptase buffer (10 X) supplemented with dNTPs (0.5 μM each), oligo (dT) primers (2.5 μM), RNase inhibitor (10 units) and Omniscript Reverse Transcriptase (Qiagen). The first strand DNA synthesis was carried out at 37 °C for 60 min. PCR amplification was performed in 50 μl volume using 5 μl PCR Buffer Hot Star (10 X), 10 μl Q Solution (5 X), dNTPs (200 μM), 0.4 μM of each corresponding primer and 2.5 units of TAQ™ DNA Polymerase (Qiagen). The PCR amplifications were performed using a GeneAmp PCR System 9700 (Applied Biosystems) under the following profile: 28-35 cycles of amplification were used at 95 °C for 30 s, 58 °C for 30 s and 72 °C for 40 s. Amplicons were visualized on standard ethidium bromide stained 1.5% agarose gel and analyzed using Scion Image software (Marcil et al. 2003).
THP-1 cell adhesion assay
HUVEC were seeded in 6-well plates 48 hours before the experiment. Only confluent HUVEC monolayers were used, as confirmed by microscopic inspection. THP-1 cells were incubated in 6 ml supplemented RPMI 1640 medium containing 10 μg/ml of the fluorescence dye 2',7'-bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) (Molecular Probes, Eugene, Oregon) at 37 °C for 20 minutes. Dye loading was stopped by addition of 44 ml RPMI 1640 with 5% FBS. Fluorescence-labeled cells were resuspended (106/ml) in supplemented EBM media without FBS. Before addition of THP-1 cells, HUVEC were washed with supplemented EBM media without FBS and then stimulated with TNF-≈ (100 U/ml) for 3 hours. A suspension of THP-1 cells containing or not iron/ascorbate and the antioxidant Trolox was added to HUVEC for an incubation of 20 minutes at 37 °C. After, the THP-1 suspension was withdrawn and thoroughly washed with PBS. Cells were lysed with lysis buffer (pH 8) and fluorescence was measured by a Luminescence Spectrometer LS 50 (Perkin Elmer Instruments, Waltham MA) (excitation, 485 nm; emission, 535 nm). Adherent cells per well were calculated by comparing the determined fluorescence to a standard curve of THP-1 labeled with BCECF-AM (Bernardo et al. 1993; Weber et al. 1994).
Statistical Analysis
Data from the experiments were analyzed with SPSS Software 11.0 using ANOVA test. Reported values are expressed as means ± SD. Statistical significance was accepted at P < 0.05.
Results
MDA generation in HUVEC after iron/ascorbate exposure
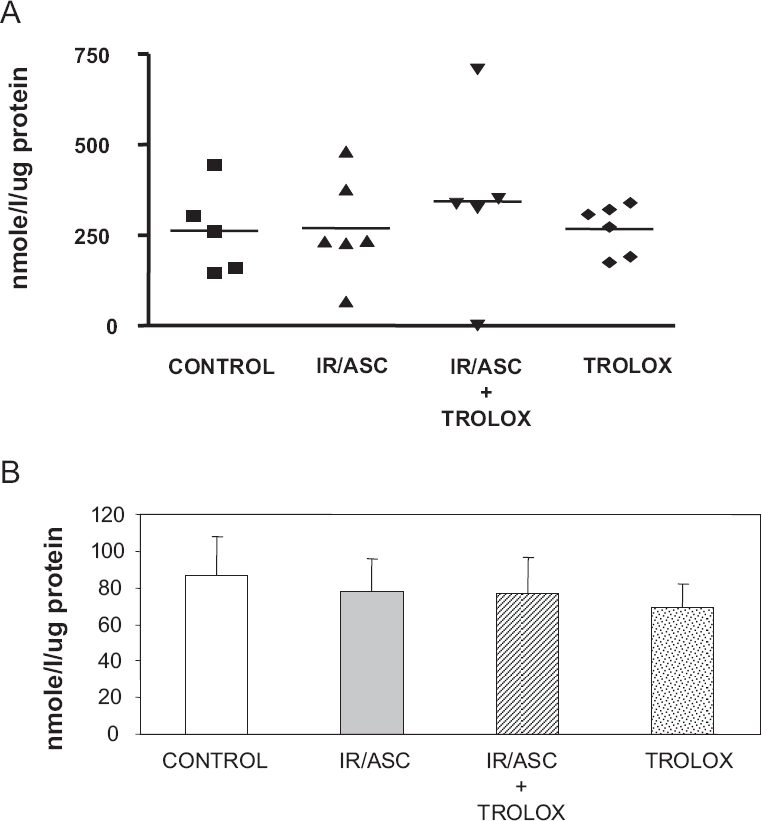
The effectiveness of iron/ascorbate in initiating lipid peroxidation was tested after incubation with HUVEC cells. At the end of a 4-h culture period, the degree of lipid peroxidation was determined by measuring MDA in cells. As illustrated in Figure 1A, iron/ascorbate promotes the production of peroxidation above control values. The formation of MDA was 27-fold higher (P < 0.001) in the presence than in the absence of iron/ascorbate. Neither iron nor ascorbate alone could induce marked lipid peroxidation (data not shown), showing that the reaction between the two is necessary to induce oxidative stress. The efficiency of powerful antioxidants in preventing or reducing lipid peroxidation induced by iron/ascorbate was also evaluated. Trolox significantly (P < 0.002) suppressed cellular peroxidation (3.5-fold) induced by iron/ascorbate at the concentration tested (Fig. 1A), without any toxic effect as shown by the MTT test (Fig. 1B). However, BHT, known as a powerful antioxidant either in ethanol or DMSO, was less efficient and even deleterious for the cell viability (Fig. 1B). Thus, Trolox was chosen for the subsequent experiments. We, therefore, selected this concentration of iron/ ascorbate and Trolox for the subsequent studies since it does not represent a pharmacological dose. Importantly, cells were incubated with iron/ascorbate for limited periods of time (4 and 8 hours) since longer exposition to iron/ascorbate led to HUVEC morphologic changes and loss of cell adherence.

Cholesterol influx and efflux in HUVEC
We next investigated if cholesterol flux is influenced by iron/ascorbate-mediated lipid peroxidation. HUVEC endothelial cells were exposed to iron/ ascorbate with or without the antioxidant Trolox for 4 h and were incubated for 2 h with 25 mg of protein/ml oxLDL-[3H]-cholesteryl ether and cholesterol influx was then measured. As shown in Figure 2A, the incorporation of oxLDL-[3H]-cholesteryl ether remained unchanged either with iron/ascorbate or antioxidant. Similarly, cholesterol efflux to HDL3 after loading cells with [3H]-cholesteryl oleate albumin and washing was not altered by iron/ascorbate (Fig. 2B). We concluded that lipid peroxidation did not affect the influx and efflux processes.
Assessment of gene and protein expression of ABCA1, SR-BI and LOX-1 in HUVEC
We attempted to define the status of lipoprotein-cholesterol receptors in HUVEC cells following exposure to iron/ascorbate. To this end, ABCA1 was characterized as the rate limiting unidirectional cellular cholesterol exporter. Similarly, we evaluated mRNA and protein mass of SR-BI that may promote the bidirectional flux of free cholesterol between HUVEC cells and lipoproteins and/or the selective uptake of esterified cholesterol, and of LOX-1 implicated in oxidized lipid uptake. Gene expression of ABCA1 was diminished by iron/ ascorbate, but only after an 8 hour-incubation period (Fig. 3A) and Trolox was able to partially, but not significantly, prevent the iron/ascorbate-induced ABCA1 fall. On the other hand, no significant modifications were noted in protein expression of ABCA1 (Fig. 3B) following the incubation of HUVEC with iron/ascorbate. Similarly, no significant alterations were recorded in mRNA and protein mass of SR-BI or LOX-1 (data not shown).

Gene expression of nuclear factors in HUVEC
We further tested PPARs and LXRs, which are nuclear receptors extensively involved in the control of lipid metabolism. The mRNA levels of PPAR≈, PPARβ/δ, PPARγ, LXR≈, and LXRβ were quantified by RT-PCR. Iron/ascorbate-mediated lipid peroxidation was unable to induce changes in these nuclear factors in comparison to controls (Fig. 4).

Gene expression of adhesion molecules in HUVEC
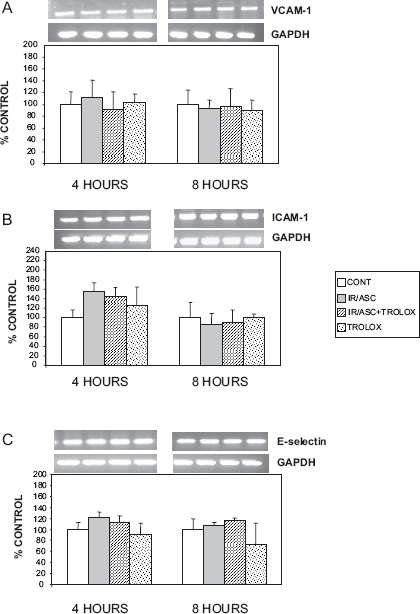
Since atherosclerosis is characterized by adhesion and transendothelial migration of monocytes, we have analyzed the gene expression of adhesion molecules that strongly promote this process. Iron/ ascorbate-mediated lipid peroxidation did not affect the status of VCAM-1, ICAM-1 and E-selectin (Fig. 5).
Iron/ascorbate-mediated changes: a comparison between HUVEC and THP-1
Comparison between HUVEC endothelial cells and THP-1 derived macrophages was undertaken to examine the effects of iron/ascorbate on cholesterol efflux/influx, lipoprotein receptors (ABCA1 and SR-BI) and transcription factors. Table 1 summarizes the findings. Changes were noted in ABCA1 protein expression, PPAR≈ and PPARγ gene expression, as well as LXR≈ and LXRβ gene expression.
Comparative Impact of Oxidative Stress.

The transporters of cholesterol flux as well as transcription factors were assessed and comparison was undertaken between HUVEC and THP-I macrophages. The results are presented as % of own control values (100%) in each experimental group. Statistical significances refer to the respective control group.
p < 0.05;
p < 0.002;
p < 0.003;
p < 0.006;
p < 0.01.
Co-culture of endothelial cells and monocytes
Interactions between endothelial cells and monocytes in the vascular wall appear to be important in determining vascular function and remodelling (Simionescu et al. 1993; Ross 1993). Since cell-cell interaction is an essential component of atherosclerotic plaque development, it was valuable to find out how oxidative stress influences contact co-culture of endothelial cells with monocytes. Figure 6 showed that iron/ascorbate-mediated lipid peroxidation augmented cell adhesion of THP-1 and HUVEC. Under these experimental conditions, there was an increase of ICAM-1, E-selectin and MCP-1 gene expression, whereas endothelial nitric oxide synthase (eNOS) mRNA was diminished and VCAM-1, P-selectin and LOX-1 remained unchanged (Fig. 7).


Discussion
Oxidative damage to LDL is known to accelerate the development of atherosclerosis through the formation of foam cells (Parthasarathy et al. 1989) and auto antibodies against oxLDL (Faviou et al. 2005). However, the effect of oxidative stress per se on contributing factors in atherosclerosis is poorly documented. We have therefore evaluated the impact of iron/ascorbate on endothelial cells and consequently reported an appreciably raised concentration of MDA without any change in cholesterol influx/efflux and lipoprotein-cholesterol receptors (such as SR-BI, ABCA1 and LOX-1), as well as the gene expression of nuclear receptors (including PPAR≈, PPARβ/δ, PPARγ, LXR≈, and LXRβ) and the level of VCAM-1, ICAM-1 and E-selectin mRNA. On the other hand, iron/ascorbate-mediated lipid peroxidation enhanced cellular adhesion of HUVEC and THP-1 along with an increase in ICAM-1, E-selectin and MCP-1 gene expression, a decline in eNOS and invariable alteration in VCAM-1, P-selectin and LOX-1.
In our studies, we used HUVEC as a model of endothelial cells given the ready availability, ease of preparation, and extensive literature on these cells. HUVEC have been employed to study a range of important pathophysiological processes, including immune-endothelial interactions, endothelial dysfunction related to atheroma formation and angiogenic sprouting, and capillary lumen formation (Nakatsu et al. 2003). In particular, HUVEC have been crucial for the investigation of endothelial cell involvement in the diverse steps of atherosclerosis development. In fact, several reports have recently emphasized the direct impact of oxLDL on the endothelium (Florian et al. 2007), vasorelaxation factors (Jantzen et al. 2007), shear stress response (Burns et al. 2007), and inflammation effectors (Rodriguez et al. 2007; Zhou et al. 2007). However, additional studies are necessary before extrapolating our findings to humans.
Both ABCA1 and SR-BI are known as apolipo-protein (apo) A-I- or HDL-binding proteins. ABCA1 has been identified as the primary gatekeeper for eliminating tissue cholesterol, since it mediates the efflux of phospholipids and cholesterol onto apo A-I (Bodzioch et al. 1999; Lawn et al. 1999; Brooks-Wilson et al. 1999). Its activity is rate limiting for HDL biogenesis in the liver and helps to maintain the cholesterol homeostasis of macrophages in the vascular wall (Van et al. 2005; Yokoyama 2005). SR-BI is an HDL receptor that can mediate selective uptake of HDL cholesteryl esters by cells (Krieger 1999) but can also promote cellular free cholesterol efflux to HDL and the reorganization of a cholesterol oxidase-sensitive pool of cellular cholesterol (Yancey et al. 2003). In contrast to other cells that participate in the formation of atherosclerotic plaque, endothelial cells remain enigmatic as to their cholesterol homeostasis. Our data confirmed the presence of ABCA1 (Hassan et al. 2006) and SR-BI (Yuhanna et al. 2001) in endothelial cells, but could not demonstrate any modification of ABCA1 and SR-BI expression by oxidative stress. Accordingly, the influx and efflux of cholesterol was not changed by oxidative stress. Therefore, our results suggest that oxidative stress differently operates in endo-thelial cells than in macrophages in view of our recent data showing that exposure of THP-1 derived macrophages to iron/ascorbate decreased cholesterol efflux and diminished the gene and protein expression of ABCA1 (Marcil et al. 2006).
LOX-1 is the major oxLDL receptor found on endothelial cells (Sawamura et al. 1997; Chen et al. 2002). LOX-1 mediates the internalization of oxLDL into cells and can induce endothelial cell dysfunction that is believed to constitute an early step in the development of atherosclerosis (Kita et al. 2001). The expression of LOX-1 enhances a variety of intracellular processes that lead to expression of adhesion molecules, to which inflammatory cells attach, and endothelial activation, which affects a variety of gene expression such as endothelial constitutive eNOS and MCP-1 (Moriwaki et al. 1998; Kita 1999; Mehta et al. 2003; Mehta 2004). However, in the present investigation, iron/ascorbate-mediated lipid peroxidation did not alter the expression of LOX-1 and did not affect the status of VCAM-1, ICAM-1 and E-selectin.
Nuclear receptors represent a large super family of ligand-dependent transcription factors that regulate the expression of target genes to affect diverse lipid-metabolic processes. Upon activation by specific ligands, LXRs and PPARs form obligate heterodimers with the retinoid X receptor (RXR) and bind to LXR or PPAR response elements located in the promoter region of their target genes. PPARs and LXRs control the transcription of a number of specific genes involved in central pathways in cholesterol metabolism, transport, and elimination (Lu et al. 2001; Marx et al. 2004). For example, oxLDL inhibited apo A-I-mediated cholesterol efflux in endothelial cells and suppressed ABCA1 at the protein and mRNA levels in HUVECs by decreasing ABCA1 promoter activity via a ligand-dependent inhibition of LXR (Zhu et al. 2005). Our observations could not reveal any modifications in the gene expression of these nuclear receptors by the presence of oxidative stress. Furthermore, our findings suggest that (i) the two cellular populations of the endothelium, endothelial cells and macrophages, are differentially regulated (Marcil et al. 2006); and (ii) the pro-inflammatory and pro-atherogenic effects of oxLDL are not observed with iron/ascorbate-mediated lipid peroxidation.
Much evidence supports a pivotal role for inflammation in all phases of atherosclerosis and the major cellular participants include immune cells. Monocytes/macrophages are critical cells present at all stages of atherogenesis and, when stimulated, can produce biologically active mediators that have a profound influence on the progression of atherosclerosis. Our studies showed that oxidative stress promotes monocyte adhesion to endothelial cells and the direct cell-cell interaction resulted in high levels of the gene expression of ICAM-1, E-selectin and MCP-1, whereas eNOS mRNA was diminished. To our knowledge, this is the first report that documents the significant contribution of iron/ascorbate mediated lipid peroxidation as a potentially injurious stimulus for the endothelium without the involvement of oxLDL. Under our experimental conditions, the concomitant presence of oxidative stress and monocytes was necessary for the induction of chemotactic factors and the down-regulation of eNOS, which may compromise the health of the endothelium in vivo situation.
Conclusion
Overall, our studies show that, contrary to THP-1 derived macrophages, cholesterol metabolism and cellular inflammatory responses are not altered by oxidative stress in HUVEC. However, in the presence of monocytes, HUVEC respond to oxidative stress in a pro-inflammatory manner, suggesting that monocyte priming in necessary to HUVEC reaction. Further studies are required to deepen endothelial cell-monocyte cross talk in order to understand the implication of oxidative stress in cardiovascular disease.
Footnotes
Acknowledgements
This work was supported by grants from Canadian Institutes of Health Research (MOP 49433), Crohn's and Colitis Foundation of Canada (CCFC) and Canadian Diabetes Association (OG-2-07-2422-EL). The authors thank Mrs Schohraya Spahis for her technical assistance.