
Abstract
Acute ischemia results in deadly cardiac arrhythmias that are a major contributor to sudden cardiac death (SCD). The electrophysiological changes involved have been extensively studied, yet the mechanisms of ventricular arrhythmias during acute ischemia remain unclear. What is known is that during acute ischemia both focal (ectopic excitation) and nonfocal (reentry) arrhythmias occur, due to an interaction of altered electrical, mechanical, and biochemical properties of the myocardium. There is particular interest in the role that alterations in intracellular calcium handling, which cause changes in intracellular calcium concentration and to the calcium transient, play in ischemia-induced arrhythmias. In this review, we briefly summarize the known contributors to ventricular arrhythmias during acute ischemia, followed by an in-depth examination of the potential contribution of altered intracellular calcium handling, which may include novel targets for antiarrhythmic therapy.
Acute Ischemia and Sudden Cardiac Death
Disturbances in cardiac rhythm resulting from an acute reduction in blood supply to a region of the heart are a major cause of sudden cardiac death (SCD).1,2 Specifically, lethal ventricular arrhythmias (sustained ventricular tachycardia [VT] or ventricular fibrillation [VF]) due to acute coronary artery occlusion account for 80% of all cases of SCD without a prior history of heart disease. 3 The electrophysiological changes leading to the induction and sustenance of ventricular arrhythmias in the acute (~1 hour) period of regional ischemia (phase 1, during which approximately 50% of all SCDs occur 4 ) have been extensively studied,5,6 yet the mechanisms remain unclear. Here, we briefly summarize known contributors to acute ischemia-induced ventricular arrhythmias, followed by a more detailed exploration of the potential role that changes in intracellular calcium (Ca2+) handling may play in that setting.
Ischemia-Induced Ventricular Arrhythmias
Ventricular arrhythmias in acute ischemia are caused by focal (ectopic excitation) and nonfocal (reentry) mechanisms, due to an interaction of alterations in myocardial electrical, mechanical, and biochemical properties (summarized below – for further details and references, see reviews by Carmeliet 5 or Janse and Wit 6 ).
Subcellular Effects
Principal ischemia-induced intracellular changes include: (i) decreased adenosine triphosphate (ATP) and increased adenosine diphosphate (ADP) (due to hypoxia/anoxia); (ii) decreased pH (resulting from carbon dioxide retention and increased proton production); (iii) increased sodium (Na+) concentration (caused by increased Na+ influx and decreased efflux); (iv) increased Ca2+ concentration ([Ca2+]i, discussed below); (v) activation of the ATP-inactivated potassium (K+) current (IK,ATP, with an increase in the ADP/ATP ratio); (vi) decreased fast Na+ current (INa, primarily a result of increased inactivation due to acidosis, oxidative stress, and depolarization of resting transmembrane potential, Vm); and (vii) decreased L-type Ca2+ current (ICa,L, discussed below). These occur at the same time as important interstitial changes, including increased extracellular K+ concentration (largely due to increased K+ efflux via IK,ATP, and to a lesser extent to decreased influx and shrinkage of the extracellular space) and elevated levels of catecholamines (due to acidosis-induced endogenous release from sympathetic nerve terminals).
Cell- and Tissue-Level Effects
At the whole cell level, extracellular hyperkalemia causes a shift in the equilibrium potential for K+, which results in depolarization of resting Vm (facilitated by an increasing inward leak current). Initially, this causes an increase in cell excitability as Vm approaches threshold. As extracellular K+ concentration continues to increase, there is a subsequent decrease in the rate of the action potential (AP) upstroke and in AP amplitude, as INa is decreased due to channel inactivation. At the same time, the AP is at first slightly prolonged (due to a decrease in local temperature of the myocardium), followed by AP shortening (primarily by activation of IK,ATP). In contrast to the effect on AP duration (APD), ischemia causes an increase in the effective refractory period (ERP, caused by INa inactivation), which results in post-repolarization refractoriness (although ERP is shortened at the border between healthy and fully ischemic tissue, explained below). In tissue, conduction velocity (CV, which is determined by AP upstroke velocity and amplitude, as well as the ease of intercellular propagation) is reduced, at first by the change in AP characteristics, and then more severely with the closure of gap junctions as ischemia progresses.
Mechanisms of Arrhythmogenesis
As acute ischemia is dynamic, with effects progressing in time after coronary artery occlusion, so is the frequency of the associated arrhythmias. Arrhythmias tend to occur in two distinct phases. Specific timings vary with species and experimental preparation, but in general the first phase (phase 1a) occurs approximately 2-10 minutes after occlusion (with a peak in arrhythmias at around 6 minutes), while the second phase (phase 1b) occurs after approximately 20-40 minutes (with a peak at around 30 minutes). In addition, effects are nonuniform across the ischemic region, with heterogeneities pronounced at the ischemic border. These heterogeneities, such as an increase in excitability and a shortening of ERP in the border zone (due to injury current), are important factors in the generation of arrhythmias.
The nature and underlying mechanisms of arrhythmias in the two subphases of acute ischemia are also distinct. Phase 1a arrhythmias are not generally lethal, manifesting as short periods of VT only (facilitated by the ischemia-induced decrease in excitation wavelength = APD X CV). These arrhythmias are mostly triggered by reentry near the border zone, with abnormal impulses traveling through an extended circuit in the ischemic tissue or multiple wavefronts around a region of conduction block (although some ectopic excitation occurs as excitability is initially increased). Phase 1b arrhythmias, on the other hand, are more deadly (and thought to be the primary source of SCD), as they often degenerate into VF. The mechanism of their genesis, however, is less clear. In phase 1b, there is a greater extent of ectopic excitation than in phase 1a, both in the ventricular myocardium and in Purkinje fibers, most likely resulting from a combination of depolarizing influences of increased catecholamine levels (stimulating, for instance, intracellular Ca2+ release), enhanced stretch of border zone tissue (which stimulates stretch-activated currents), an increased source–sink relationship (due to cellular uncoupling), increased [Ca2+]i, and increased injury current. At the same time, the propensity for triggered activity to result in reentry is heightened by the decrease in CV (as gap junctions begin to close) and a further increase in extracellular K+ concentration. As mentioned earlier, the extensive reviews by Carmeliet 5 or Janse and Wit 6 can be referred to for references and further details.
Changes in Intracellular Calcium Handling during Acute Ischemia
While it is clear that many factors contribute to arrhythmias during acute ischemia, the involvement of altered intracellular Ca2+ handling is of particular interest, as it appears to have a critical role in arrhythmogenesis in various other disease states. 7 Specifically, acute ischemia is known to affect various aspects of Ca2+ handling, resulting in important changes in [Ca2+]i and the Ca2+ transient.
Intracellular Calcium Handling under Physiological Conditions
Under normal conditions, excitation rapidly propagates into the cell interior via specialized sarcolemmal invaginations (transverse tubules). Depolarization causes opening of voltage-sensitive L-type Ca2+ channels (LCCs) concentrated in this region, which causes an influx of Ca2+ into the dyadic cleft and a localized increase in [Ca2+]i. In direct apposition to LCCs are sarcoplasmic reticulum (SR) release channels (ryanodine receptors [RyR]), which together form couplons that allow a regional increase in [Ca2+]i to stimulate RyR opening and cause release of Ca2+ from the SR (a process known as Ca2+-induced Ca2+ release [CICR]). This large increase in [Ca2+]i (up to 10 times diastolic levels) forms the upstroke of the Ca2+ transient, during which Ca2+ binds to troponin C on the myofilaments, enabling cross-bridge cycling and muscle contraction. Upon relaxation, Ca2+ is released from the myofilaments and then either extruded from the cell by the Na+-Ca2+ exchanger (NCX, ~28% of cytosolic Ca2+, and to a lesser degree the sarcolemmal Ca2+ ATPase, ~2%), or resequestered in the SR by the SR Ca2+ ATPase (SERCA, ~70%), so that preexcitation Ca2+ concentrations are restored. It is worth noting that only a small fraction of cytosolic Ca2+ is free at rest, with most bound to cytoplasmic proteins.
Ca2+ is also found in other intracellular compartments, where it is has important regulatory functions. In the mitochondria, Ca2+ regulates dehydrogenase activity to match energy production to utilization. The free concentration of Ca2+ in the mitochondria is generally lower than in the cytosol and follows changes in that compartment. Ca2+ influx occurs primarily through the uniporter driven by an electrical gradient (the mitochondrial matrix is negative with respect to the cytoplasm). Efflux depends on the proton gradient, occurring through mitochondrial NCX and the Na+/hydrogen (H+) exchanger (NHE), which act in concert to keep intramitochondrial Ca2+ and Na+ concentrations at low levels. As the mitochondria constitute a large fraction of cell volume, they can accumulate a relatively large amount of Ca2+, and as such act essentially as a Ca2+ buffer. Specific details regarding normal intracellular Ca2+ handling can be found in the comprehensive review by Bers 8 or Santulli and Marks. 9
Ischemic Effects on Calcium-Induced Calcium Release
Effects on L-Type Ca2+ Channel Activity
It has been shown in human embryonic kidney cells expressing the α1-subunit of the LCC (which contains the pore-forming and voltage-sensing regions of the channel and is able to conduct ions in the absence of other accessory subunits) that hypoxia alone impairs ICa,L activation kinetics in a PO2-dependent manner. 10 This is thought to occur via posttranslational redox modification of the channel 11 or a change in the sensitivity to changes in oxygen tension, 12 although confirmation of their relative contribution is still needed. 13 With reduced oxygen availability, there is a reduction in ATP, which further affects ICa,L by decreasing peak current, as demonstrated in isolated guinea pig ventricular myocytes. 14 On the other hand, it has also been shown in these cells that hypoxia increases LCC sensitivity to β-adrenergic stimulation (as occurs with increased catecholamine levels during acute ischemia), which acts to increase ICa,L, opposing the reduction in activation and peak current. 11 In the case of combined oxidative and glycolytic metabolic inhibition (as occurs in phase 1b), the efficacy of LCCs to initiate CICR is impaired, causing a reduction in the ratio of ICa,L to Ca2+ transient amplitude. 15 This effect, however, was found not to be linked with LCC dephosphorylation, so may be a result of alterations in RyR activity.
Acidosis also affects IC,aL, reducing peak current in isolated guinea pig ventricular myocytes. 16 In isolated rabbit hearts, reductions in pH to 6.3 have been shown to produce a ~70% reduction in the maximal conductance of ICa,L. 17 This effect is enhanced when combined with inhibition of the NHE, 18 as occurs in phase 1b. Of interest, in isolated rabbit sinoatrial node cells, ischemia in fact increases ICa,L, 19 which is thought to relate to differences in the LCC α-subunit found in sinoatrial node (Cav 1.3) vs. ventricular (Cav1.2) cells. 20 In addition, there is a reduction in the conductance of T-type Ca2+ channels during acute ischemia, 19 but the expression of that channel in ventricular myocytes is negligible. 5
Ligands produced during acute ischemia similarly affect ICa,L. Amphiphiles (long-chain acylcarnitines, LCAs) accumulate in ischemia as β-oxidation slows. 21 The effect of LCAs on LCCs is debated, with previous literature indicating both current amplification 22 and inhibition. 23 In support of the importance of an LCA effect on Ca2+ handling during ischemia, however, is the finding that in isolated rat ventricular myocytes, an increase in LCAs is associated with afterdepolarizations (although this may also be linked to enhanced INa). 24 Reactive oxygen species (ROS) have also been shown to be produced during acute ischemia in cultured chick embryo ventricular myocytes25,26 and intact guinea pig hearts. 27 ROS is produced with mitochondrial electron transport chain damage 28 or uncoupling of nitric oxide synthase (NOS), 29 and by NADPH 30 or xanthine 31 oxidase (for further details regarding ROS production during ischemia, see the review by Raedschelders et al. 32 ). These volatile molecules have been shown to increase ICa,L density, 33 via redox modification of the LCC α1-subunit.34,35 LCCs are also sensitive to the increase in [Ca2+]i that occurs with ischemia, which increases the rate of their inactivation (via modulation of the α1-subunit by the Ca2+-calmodulin complex 36 ), resulting in a negative inhibition of LCCs (similar to the voltage dependence of their inactivation). 37
Effects on RyR Activity
RyR channels are regulated by various accessory proteins and their modulators, including cytosolic calmodulin, FK506-binding protein (FKBP 12.6), protein kinase A, protein phosphatases, and luminal triadin, junctin, and calsequestrin. 8 Within the first hour of ischemia, it has been shown in canine myocardium and isolated rat hearts that the number of available RyR channels is significantly reduced.38,39 The sensitivity of the RyR to cytosolic Ca2+ is reduced by progressive acidification, and to a lesser extent reductions in ATP, 40 which is an important modulator of RyR sensitivity to both luminal and cytoplasmic SR [Ca2+].41,42 Conversely, increased ROS, which alters the oxidative status of RyR, leads to enhanced Ca2+ sensitivity and leak. 30 This posttranslational modification of RyR by ROS has been linked to an increase in the open probability of the channel by S-glutathionylation. 43 Furthermore, the activity of NOS has been shown to be augmented early in acute ischemia,44,45 promoting an increase in nitric oxide and cGMP production. 46 This increase in NOS activity, acting through cGMP-dependent or -independent pathways, modifies phosphodiesterase activity or nitrosylation of cysteine residues on proteins, 47 which reduces ICa,L,48–50 while amplifying RyR and SERCA activity (thus increasing Ca2+ spark frequency).48,51
Ischemic Effects on Calcium Extrusion and Reuptake
Effects on NCX Activity
With a reversal potential of ~-35 mV, NCX typically functions during diastole in forward mode, extruding one Ca2+ ion in exchange for three Na+ ions, and thus generating a net inward current. 52 During ischemia, however, it has been proposed that NCX begins to function primarily in reverse mode, reducing Ca2+ efflux while increasing influx, thus contributing to cytosolic Ca2+ overload. 53 This is thought to be linked to the increase in intracellular Na+ concentration resulting from enhanced NHE activity in response to the reduction in intracellular pH.54,55 There is some debate, however, regarding the changes in NHE activity that occur with ischemia, as some have shown that NHE is inhibited by ischemia. 18 Inhibition of NHE is thought to be driven by a decrease in extracellular pH, 56 but others have shown that the stimulatory effects of low intracellular pH dominate. 57 Either way, as ischemia progresses, NCX function is generally reduced. 58
Effects on SERCA Activity
During acute ischemia, SERCA expression has been shown to be relatively unchanged. 59 Its function, however, is highly dependent on metabolic status (decreased ATP in ischemia reduces the energy available for its activity), in addition to the activity of the inhibitory protein phospholamban (PLB). 60 Within the first ~20 minutes of ischemia, PLB has been shown to increase phosphorylation of SERCA. 61 This increase in phosphorylation results from reduced activity of protein phosphatases by acidotic inhibition, thus maintaining the phosphorylation status of PLB and of SERCA activity. 62 As ischemia progresses, the maximum rate of SERCA conductance is reduced,63,64 and dephosphorylation of PLB by calcineurin-activated protein phosphatases impairs SERCA kinetics and reduces SR Ca2+ load. 59 Redox modification of SERCA has also been demonstrated, however, its effects are generally limited. 65
Ischemic Effects on Intracellular Calcium Concentration and Calcium Transient Morphology
During acute ischemia, Ca2+ influx begins to exceed efflux (with some delay), due to reduced efficiency of Ca2+ removal by the NCX (and ultimately influx when it operates in reverse mode),54,55 combined with an increased inward leak current. This causes diastolic [Ca2+]i to increase, plateauing at approximately three times that of initial values by 30 minutes.17,66,67 This increase in [Ca2+]i is facilitated by the ischemia-induced reduction in Ca2+ uptake by the SR,63,64 as well as displacement of Ca2+ from cytoplasmic binding sites by protons, 68 an increase in Ca2+ spark frequency, 69 and decreased myofilament Ca2+ sensitivity. 70 At the same time, the increase is mitigated by accumulation of Ca2+ in mitochondria due to reduced mitochondrial electrical and proton gradients as oxidative metabolism is reduced. 71
Ischemia-induced alterations in CICR, on the other hand, result in a decrease in SR Ca2+ release, which manifests as a decrease in the rate of the Ca2+ transient upstroke 72 and in Ca2+ transient amplitude. 73 At the same time, the rate of Ca2+ transient decay is decreased, 74 although Ca2+ transient duration is generally maintained (or only slightly increased).72,75,76 In the face of a decreasing APD, a maintained (or lengthened) Ca2+ transient is partly accounted for by slowed RyR kinetics, which reduces the rate of Ca2+ release, thus indirectly delaying recovery, 77 which is enhanced as SERCA function declines. Yet this appears to occur without a change in SR Ca2+ content, 17 suggesting that ischemia equivocally impairs both release and reuptake mechanisms.
Calcium-Driven Arrhythmias during Acute Ischemia
Ischemia-induced changes in intracellular Ca2+ handling may have important implications for arrhythmogenesis, contributing to arrhythmic triggers (premature excitation) and substrate (proarrhythmic condition creating a setting for reentry). 78
Arrhythmic triggers driven by Ca2+, manifesting as premature depolarizations, are commonly classified as early or delayed afterdepolarizations (EADs or DADs, respectively). 79 EADs occur during the plateau or repolarization phase of the AP. In many disease states, EADs during the AP plateau occur when repolarization is delayed, resulting in an increase in APD sufficient for the reactivation of ICa,L and a subsequent depolarizing current.80,81 During acute ischemia, however, APD is shortened and ICa,L inhibited, so it is unlikely that EADs occur during the AP plateau in that setting. EADs during AP repolarization, on the other hand, have been shown to occur both with increased and decreased APD. In the case of increased APD, EADs during AP repolarization can occur independent of changes in cytosolic Ca2+, resulting from electrotonic interactions between regions of varying APD,80,82 which may be important during regional ischemia, 83 where spatial heterogeneities in APD exist between ischemic and healthy tissues.84,85 In the case of shortened APD, on the other hand, if Ca2+ transient duration in maintained (as is the case during much of the acute phase of ischemia), [Ca2+]i remains elevated during cellular repolarization (under normal conditions, AP and Ca2+ transient duration are generally similar 86 ). This divergence of AP and Ca2+ transient duration creates a vulnerable window for EADs to occur, due to the depolarizing current generated by the NCX,87,88 which is exasperated by the increased intracellular Na+ concentration. 89 Evidence for the importance of this mechanism in acute ischemia comes from a global ischemia model in the isolated rabbit heart with Ca2+ chelation by BAPTA. 76 In that study, it was shown that during global ischemia AP duration decreased, while Ca2+ transient duration increased (Fig. 1A and C). With pre-application of BAPTA, however, Ca2+ transient duration instead decreased during ischemia (and to a similar degree as AP duration; Fig. 1B and D). Thus, while the duration of the Ca2+ transient was greater that the AP in both cases, the difference was higher without BAPTA (Fig. 1C and D), such that Ca2+ chelation reduced the period over which the Ca2+ transient intruded into electrical diastole (Fig. 1A and B). This change was associated reduction in the incidence of endocardial focal discharges (a result attributed to a reduced incidence of EADs).
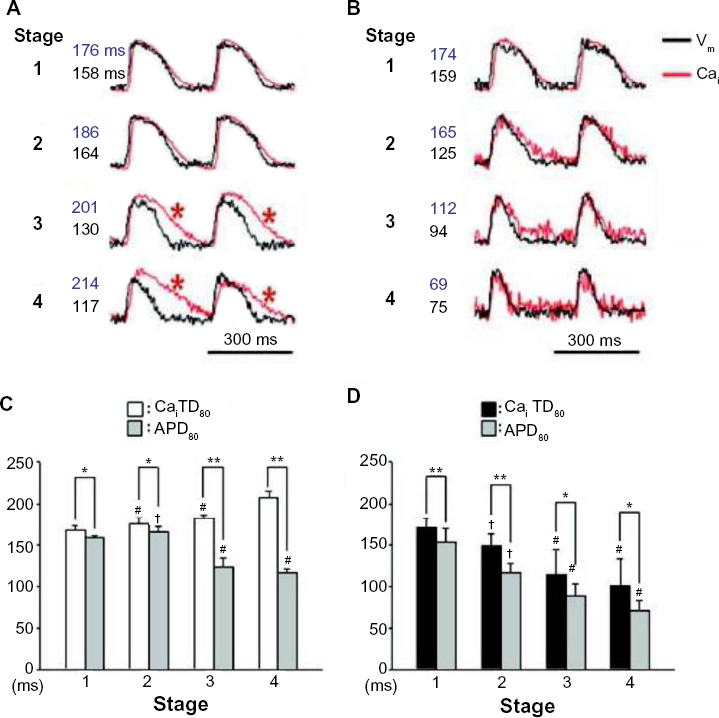
Evidence for the importance of a difference in calcium transient duration (CaiTD80) and action potential duration (APD80) for early afterdepolarizations in acute ischemia. (
DADs, on the other hand, occur during the resting phase of the AP. The driving current for DADs has been shown in both isolated human ventricular myocytes and sheep ventricular and Purkinje cells to be generated by elevated [Ca2+]i, acting through the NCX 90 or the Ca2+-activated chloride channel. 91 This has been demonstrated to be an important mechanism for ischemia-induced arrhythmias in chronically instrumented dog experiments, in which pretreatment with BAPTA reduced the incidence of arrhythmias. 92 Moreover, it has been suggested that both DADs and EADs during ischemia may be more prevalent in endocardial tissue,76,93 potentially relating to transmural differences in SERCA expression 94 and [Ca2+]i. 95
EADs and DADs may be exasperated by additional factors during acute ischemia. Nonuniform contraction that occurs with changes in mechanical properties of the myocardium, especially stretch at the border zone, may contribute to EADs and DADs. 96 While stretch-activated channels may partly account for stretch-induced premature excitation,97,98 stretch also causes significant changes in intracellular Ca2+ handling.99–101 Stretch can result in an acute and transient increase in Ca2+ spark frequency, 102 related to increased ROS. 103 At the same time, stretch increases the affinity of troponin C for Ca2+, such that with stretch more Ca2+ is bound and then released upon relaxation, which slows Ca2+ transient decline 104 and can result in Ca2+ waves, 105 DADs, 106 and sustained arrhythmias. 107 The localized increase of catecholamines during acute ischemia108,109 may also have important implications for Ca2+-driven arrhythmias.110,111 β-adrenergic stimulation increases ICa,L, 112 especially during ischemia, as translocation of β-adrenergic receptors to the sarcolemma is increased 73 and the sensitivity of LCC to β-adrenergic stimulation is enhanced, 11 promoting their reactivation. Importantly, Ca2+-driven membrane depolarization becomes especially relevant under conditions of moderate cellular uncoupling during acute ischemia, 113 as the electrotonic influence of healthy myocardium on ischemic tissue (the so-called source–sink relationship) is reduced,114,115 thus facilitating the propagation of EADs and DADs. The interaction of β-adrenergic-induced Ca2+-driven afterdepolarizations and cellular uncoupling has been demonstrated with localized injection of norepinephrine in the isolated rabbit heart. 116 In that study, norepinephrine injection caused focal ventricular excitation (Fig. 2A(ii)), associated with a decrease in Ca2+ transient duration (compared to saline injection-induced excitation; Fig. 2B(i), B(ii), and D), along with a localized reduction in the Vm-Ca2+ delay (Fig. 2C(i), C(ii), and E). Under conditions of moderate cellular uncoupling (by pre-application of carbenoxolone), the effect of norepinephrine injection on Ca2+ transient duration was similar (Fig. 2B(iii) and D), however the spatial extent of changes in the Vm-Ca2+ delay was increased (Fig. 2C(iii) and E). Thus, with uncoupling, a larger effect on Vm was observed for the same effect on Ca2+, indicating a decrease in the electrotonic influence of the surrounding myocardium, which increased the propensity for Ca2+-mediated focal arrhythmias (Fig. 2F and G). As ischemia progresses, however, cellular uncoupling becomes more severe as connexin phosphorylation is reduced, in part due to the increase in [Ca2+]i117,118 (which in fact can be prevented by reducing Ca2+ overload 119 ). While this increase in uncoupling is likely to prevent propagation of EADs and DADs, it will increase the potential for reentry by causing conduction block or by slowing conduction and increasing the excitable gap. 120

Effects of localized catecholamine (norepinephrine, NE) application in the isolated rabbit heart. (
Alterations in Ca2+ handling during ischemia have also been shown to give rise to rate-dependent beat-to-beat variations in the magnitude of Ca2+ release (Ca2+ transient alternans, which occurs when excitation precedes recovery of the previous Ca2+ transient, resulting in reduced Ca2+ release, followed by a compensatory increase).73,75,121,122 Ca2+ transient alternans may be arrhythmogenic if beats with augmented Ca2+ release are sufficient to initiate afterdepolarizations.123,124 As for EADs and DADs, Ca2+ transient alternans appear to occur more readily in the endocardium, because of transmural heterogeneities in Ca2+ handling. 125 Moreover, if Ca2+ transient alternans in turn contributes to electrical alternans, the increase in dispersion of refractoriness may promote conduction slowing or block, facilitating reentry. 126
Conclusion
Arrhythmias during acute ischemia result from a combination of electrical, mechanical, and biochemical changes in the heart. Alterations in intracellular Ca2+ handling, specifically, may play an important role in both ectopic excitation and reentrant activity. With further investigation, especially focusing on the importance of intrasubject heterogeneities and intersubject variability, 127 facilitated by a combined experimental–computational approach, 128 there is the potential for identification of novel Ca2+ handling-related targets for antiarrhythmic therapy.
Author Contributions
Wrote the paper: PB, TAQ. Both the authors reviewed and approved the final manuscript.