
Abstract
Atherosclerosis is the leading cause of death in the United States and worldwide, yet more men die from atherosclerosis than women, and at a younger age. Women, on the other hand, mainly develop atherosclerosis following menopause, and particularly if they have one or more autoimmune diseases, suggesting that the immune mechanisms that increase disease in men are different from those in women. The key processes in the pathogenesis of atherosclerosis are vascular inflammation, lipid accumulation, intimal thickening and fibrosis, remodeling, and plaque rupture or erosion leading to myocardial infarction and ischemia. Evidence indicates that sex hormones alter the immune response during atherosclerosis, resulting in different disease phenotypes according to sex. Women, for example, respond to infection and damage with increased antibody and autoantibody responses, while men have elevated innate immune activation. This review describes current knowledge regarding sex differences in the inflammatory immune response during atherosclerosis. Understanding sex differences is critical for improving individualized medicine.
Introduction
Atherosclerosis is characterized by a buildup of atheromas or fibrofatty plaques in large-to-medium-sized cardiac arteries that may rupture or erode and compromise blood flow to the heart (and other organs), resulting in ischemic heart disease (IHD). The key processes in the pathogenesis of atherosclerosis are vascular inflammation, lipid accumulation, intimal thickening and fibrosis, arterial stiffness, remodeling, and plaque rupture (or erosion), leading to myocardial infarction (MI) and ischemia. Pathology progresses silently for many years or decades before clinical disease becomes apparent. The major consequences of atherosclerosis are MI (a heart attack), cerebral infarction (stroke), aortic aneurysms, and peripheral artery disease. Atherosclerosis is the leading cause of death in men and women in the United States and worldwide.1,2 Yet more men die from atherosclerosis or coronary artery disease (CAD) than women and at a younger age (40-60 years of age).2–4 Women, on the other hand, mainly develop atherosclerosis following menopause, resulting in more women “living” with atherosclerosis than men.5,6 Although women are much less likely to develop atherosclerosis than men, women with an autoimmune disease are at an increased risk of developing CAD. 7 Atherosclerosis in women is strongly associated with hypertension, or high blood pressure, and certain autoimmune diseases like systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and systemic sclerosis,2,7,8 suggesting that the immune mechanisms that lead to CAD in men are different from those in women.
Women are reported to have higher death rates following MI than men.6,9 This higher death rate is due, at least in part, to reasons that are not directly associated with the pathogenesis of atherosclerosis and include more complications following surgery in women,10,11 drugs that are more effective in men but may not be as effective or have unexpected side effects in women,5,12 and vascular assist devices (VAD) that may “fit” the chest cavity of men better than women and cause fewer side effects in men.13,14 Another reason why women die more frequently following MI may be due to misdiagnosis of atherosclerosis and a correspondingly later treatment in women. 15 A number of studies confirm that women and men differ in their reported symptoms of CAD.16,17 Although chest pain is a good predictor of obstructive CAD in men, it is not in women.15,18 Additionally, atherosclerosis occurs more often in women with apparently “normal” or nonoccluded coronary arteries.3,19–22 Since atherosclerosis, plaque rupture or erosion, and MI are inflammatory processes, these observations suggest that key differences exist in the inflammatory infiltrate between men and women with CAD. This review highlights current knowledge on sex differences in cardiac inflammation during inflammatory cardiovascular diseases (CVDs) like atherosclerosis.
Inflammation and Atherosclerosis
The key processes in the pathogenesis of atherosclerosis are vascular inflammation, lipid accumulation, intimal thickening and fibrosis, remodeling, and plaque rupture or erosion, leading to MI and ischemia. 23 Most of our understanding on the role of inflammation in atherosclerosis comes from animal models. 24 A number of recent reviews describe the role of inflammatory mediators in the pathogenesis of atherosclerosis.25–30 The atherosclerotic plaque is located within the lining of vessel walls and consists primarily of macrophages, mast cells (MCs), T cells, B cells, and dendritic cells (DCs) (Fig. 1). Plaques develop a necrotic core of lipids and a fibrous cap of smooth muscle cells and collagen. In advanced stages of disease, immune cells may also accumulate on the lumen side of the plaque, disrupting blood flow. Elevated levels of circulating cholesterol and apolipoprotein B (ApoB) undergo oxidative modifications to form oxidized low-density lipoprotein (oxLDL) that is internalized by macrophage scavenger receptors to create macrophage foam cells within plaques. 31 Most foam cells arise from mononuclear phagocytic cells, but vascular endothelial cells and smooth muscle cells can also contain lipids.25,32,33 Internalization of oxLDL results in the formation of cholesterol crystals that activate the inflammasome, leading to secretion of mediators including interleukin (IL)-1, IL-6, IL-18, chemokines, eicosanoids, proteinases, and oxidases that promote plaque inflammation, fibrosis, and remodeling.23,30,34 The inflammasome is part of the body's innate pathogen pattern recognition system, which includes nucleotide binding oligomerization domain (NOD)-like receptors and Toll-like receptors (TLRs). TLR4 activation by certain bacteria, viruses, and other agents like cholesterol crystals increases pro-IL-1β and pro-IL-18 levels, which are cleaved by the inflammasome component caspase-1 to the active form of IL-1β and IL–18. MCs and macrophages are key immune sites of inflammasome activation.

Role of inflammation in an atherosclerotic plaque. Inflammation within the vessel wall of an atherosclerotic plaque consists primarily of macrophages, mast cells, T cells (Th1, Th2, and Th17), B cells, and dendritic cells (not shown). Plaques develop a necrotic core of lipids and a fibrous cap of smooth muscle cells and collagen. In advanced stages of disease, immune cells may also accumulate on the lumen side of the plaque, disrupting blood flow and contributing to clot formation. Elevated levels of circulating cholesterol and apolipoprotein B undergo oxidative modifications to form oxidized low-density lipoprotein that is internalized by macrophage scavenger receptors to create macrophage foam cells within plaques. Internalization of oxLDL results in the formation of cholesterol crystals that activate TLR4 and the inflammasome, leading to secretion of mediators including proinflammatory and profibrotic cytokines, chemokines, eicosanoids, proteinases, and oxidases that promote plaque inflammation, remodeling, and the eventual formation of a thrombus leading to a MI. Autoantibodies against oxidized lipids (ox) may further promote disease.
Activation of TLR2, TLR4, MyD88, and the inflammasome (ie, NALP3) leads to NFκB activation and the production of IL-1β and IL-18, two proinflammatory cytokines important in the pathogenesis of atherosclerosis.30,35 Deletion of either of these cytokines in mouse models of atherosclerosis significantly reduces disease. CD4+ T helper (Th)1-type responses involving IL-18 and interferon (IFN)γ increase inflammation in plaques, and both CD4+ and CD8+ T cells are present in atherosclerotic plaques of patients.27–30 CD8+ T cells are usually associated with Th1-type immune responses. IL-18 is a strong inducer of IFNγ and drives Th1 responses, and so it should be kept in mind that a dominant Th1 response in atherosclerosis could be due to inflammasome activation in addition to the classic IL-12/STAT4-induced IFNγ pathway. Contradictory results have been found in animal models regarding the role of Th2 (ie, IL-4, IL-33) and Th17 responses in disease pathogenesis.23,36 For example, atherosclerosis is promoted in animal models by MCs, eosinophils, and/or IgE responses that require a Th2-type immune response, IL-4 and/or IL-33.37–39 In contrast, injection of mice with IL-13 or helminth antigens to induce a Th2-type immune response associated with alternatively activated M2 macrophages (ie, M2a) reduces atherosclerosis.40,41
Macrophages
Macrophages are the dominant infiltrate within the atherosclerotic plaque and follow a similar nomenclature as Th cells with M1 being driven by Th1-associated cytokines and M2 driven by Th2-associated cytokines.26,29,42 Similar to Th cells, in a typical immune response, macrophages shift from M1 to M2 during an acute response in order to regulate inflammation. 30 Importantly, a number of M2 phenotypes have been identified (ie, M2a, M2b, M2c, M2d, M4) that can be anti-inflammatory (ie, M2a, M2c) or may promote remodeling and fibrosis (ie, M2b).29,42 Although M1 macrophages are an important component of atherosclerotic plaques in mice and humans, it is important to realize that IFNs and Th1-type immune responses strongly inhibit fibrosis and remodeling by transcriptionally downregulating profibrotic cytokines like IL-4 and transforming growth factor (TGF)β1 via STAT1.43,44 Additionally, TLR3, TLR7, and TLR9 signaling, which drive dominant Th1 responses following infection, has been found to decrease atherosclerosis in animal models. 45 This means that although M1 macrophages are associated with elevated inflammation they may additionally protect against plaque remodeling, rupture, and MI. Of all of the M2 subtypes, M2b are most closely associated with IFNγ/Th1 responses because they express TLR2 and TLR4 and activate the inflammasome releasing IL-18, which elevates IFNγ and Th1 responses.29,42 Thus, macrophage subtypes that express TLR2 and/or TLR4 (ie, M2b) promote remodeling and increase the risk for MI by releasing profibrotic mediators like IL-1β and TGFβ1. IL-10, which is released from M2 and regulatory T-cell populations, inhibits inflammation but promotes foam cell formation and so its role in atherosclerosis remains somewhat ambiguous although most evidence from animal models suggests that IL-10 protects against atherosclerosis by reducing inflammation. 46
Mast Cells
Mast cells are found in atherosclerotic plaques and increase in number and degranulation at sites of plaque rupture.47–50 Degranulating MCs release profibrotic and prothrombotic factors associated with plaque rupture, thrombosis, and MI such as chymase, tryptase, IL-1β, TGFβ1, and matrix metalloproteinases (MMPs).38,44,51–53 MC-deficient mice have been found to have reduced atherosclerosis in animal models, suggesting that MCs promote CAD.37,38
Not only do MCs contribute to atherosclerosis by releasing mediators that increase inflammation, promote remodeling, and increase the risk for plaque rupture but they have also been found to release renin and activate the local renin–angiotensin system (RAS). 54 Circulating levels of angiotensinogen, released to the bloodstream from the kidneys, is converted to biologically inactive angiotensin I (AngI) by the protease renin that is released from the liver and then activated to produce AngII by angiotensin-converting enzyme (ACE) located locally on the vascular endothelium. 55 Circulating AngII via its receptors regulates blood pressure, plasma volume, and sympathetic neural activity and increases atherosclerotic plaque instability by altering MMPs among other roles.54,56,57 Renin, the rate-limiting step in the RAS, is also produced locally within the heart by cardiac myocytes, fibroblasts, endothelial cells, and MCs.54,58–60 Mast cells also release chymase and cathepsin-D, which are able to activate AngII. 54 Locally released AngII contributes to arrhythmias and acute heart failure by promoting norepinephrine release from cardiac sympathetic nerves. Blocking renin release from MCs using inhibitors or knockout mice decreases arrhythmias in animal models,61,62 suggesting that MCs play an important role in RAS-mediated promotion of atherosclerosis and heart failure. Thus, MCs are key immune cells capable of promoting multiple stages of the pathogenesis of atherosclerosis that lead to MI and heart failure.
T cells
As mentioned previously, atherosclerotic plaques of patients contain T cells. 33 In mouse models and patients, CD4+ T cells predominate over CD8+ T cells and display a dominant Th1-type immune phenotype. 47 Th1- and Th17-type responses have been found to increase atherosclerotic inflammation in animal models.36,41,63,64 In contrast, a Th2-type immune response has been found to reduce atherosclerosis. 40 Similar to M2 macrophages, regulatory T cells that release IL-10 and TGFβ appear to limit atherosclerotic inflammation.65,66 These findings highlight the difficulty in determining the role of particular immune cells and cytokines in the pathogenesis of disease, as both M2 and regulatory T cells as well as certain cytokines like IL-4 and TGFβ reduce plaque inflammation and also promote remodeling and fibrosis.
B cells and autoantibodies
B cells are also present in the atherosclerotic plaques of patients. Similar to autoimmune diseases, atherosclerosis patients have autoantibodies against LDL, oxLDL, collagen, and heat-shock proteins (eg, see Refs67–70). T-cell clones reactive to LDL have been isolated from human plaques and circulating LDL, and oxLDL autoantibodies are abundant in atherosclerosis patients. 23 In one study, autoantibodies against oxLDL discriminated between atherosclerotic and control patients better than other lipoproteins such as total cholesterol or LDL levels. 71 A correlation exists between oxLDL autoantibody levels and the extent of atherosclerosis, cardiovascular complications, and the risk of restenosis following angioplasty. 67 Elevated oxLDL/β2-glycoprotein-I autoantibody immune complexes (ICs) are associated with more severe CAD and predict a 3.5-fold increased risk for adverse outcomes, including arterial thrombosis. 72 Deposition of autoantibodies and/or ICs at the plaque site may precipitate thromboembolism.67,68 These findings indicate that autoantibodies and autoreactive T and B cells participate in the pathogenesis of atherosclerosis.
Although collagen-responsive autoreactive T cells have been found to increase Th17 responses during atherosclerosis in patients and mice, 70 the role of collagen autoantibodies remain unclear. Collagen epitopes have been found to cross-react with coxsackievirus, and it has been hypothesized that these cross-reactive autoantibody ICs may promote myocarditis and valvular heart disease. 73 Another possibility is that autoantibodies against collagen or heat-shock proteins are produced in an attempt to reduce the levels of these proinflammatory and profibrotic mediators, and so may protect and/or inadvertently promote disease. Collagen released during vessel damage or remodeling is known to activate platelets, for example, which may promote thrombosis and MI at plaque sites.74,75
Although B cells have been found to promote atherosclerosis in mice, 76 recent studies suggest that certain autoantibodies (eg, B1 cells, native ApoB immunoglobulin (Ig)M, and IgG antibodies) protect against atherosclerosis, while others (eg, B2 cells, ox-LDL IgG antibodies) promote disease. 47 Regulatory B cells as well as intravenous administration of total IgG (IVIg) have been found to reduce atherosclerosis in mice.65,67,77,78 Thus, some B cells and antibodies/autoantibodies (such as ICs) promote atherosclerosis and thrombosis while others inhibit disease, similar to the duality found for macrophages and T cells.
Sex Hormones and Cardiac Inflammation
The higher incidence and severity of atherosclerosis in men than women across all age groups suggests that sex hormones play a major role in the pathogenesis of disease.6,79 Sex differences in the incidence of CVD are also influenced by gender differences in cardiovascular risk factors and presentation of disease.80,81 That the incidence of CAD increases in women as estrogen declines with age and following menopause suggests a protective role for estrogen in the heart. However, the benefit of estrogen therapy following menopause has been controversial.82–86 Interpreting results from studies has been complicated by the mixture of hormones used for therapy (ie, estrogen and progesterone), their doses, and the age and duration (ie, timing of initiation) of therapy.6,87,88 If hormone therapy is started during or shortly after menopause, it appears to reduce the risk for CAD, 88 although more research is needed.
Underlying the sex difference in incidence of atherosclerosis is the knowledge that sex differences exist in normal heart physiology and function. For example, men have larger hearts (greater left ventricular mass), cardiac contractility is greater in healthy women than age-matched men, myocardial mass is better preserved in women as they age, cardiac apoptosis is threefold higher in men compared to women as they age, women have smaller coronary vessels than men, and premenopausal women have lower blood pressure but a faster resting heart rate than men.89–91 Sex differences in cardiac pathology also occur with aging. For example, hypertrophy, apoptosis, and fibrosis are greater in the aging male than female heart. 92 These underlying sex differences in cardiac physiology directly influence the pathogenesis of atherosclerosis.
Sex Hormones and Receptors
Estrogen, progestin, and androgen hormones act through ligand-dependent, ligand-independent, genomic, and non-genomic mechanisms. 6 The canonical pathway of hormone action involves steroid binding to an intracellular receptor that leads to dimerization and binding of the dimer complex to the regulatory element of the promoter region of specific genes. These are termed estrogen response elements (EREs) and androgen response elements (AREs) and are capable of turning on or off hundreds of genes at a time and behave essentially as ligand-activated transcription factors. This is likely the reason why such large sex differences occur for most inflammatory diseases (ie, 20:1 female to male for thyroiditis and 9:1 for SLE, for example). 93 Hormone receptors are also expressed on the surface of immune and other cells, where they mediate rapid signaling.94–96 Estrogen receptors (ERs) include ERα, ERβ, and GPR30. Each of these receptors can be found within cells and on the cell surface. ERα and ERβ are the product of two difference genes, ESR1 and ESR2, but share a high degree of homology. 6 Interestingly, a single androgen receptor (AR) is transcribed from a gene located on the X chromosome. 97
And not surprisingly, since hormones are essentially growth factors required for normal cell growth and maintenance, ERα/β, progesterone receptors (PRs), AR, and aromatase (the enzyme that converts androgens to estrogens) are expressed on/in vascular endothelial cells, vascular smooth muscle cells, cardiac fibroblasts, and cardiomyocytes in humans and rodents.6,98 Women have higher ER expression in their arteries than men, which decreases with age and menopause. 79 Estrogen via ERβ signaling has been shown to regulate arterial tone and blood pressure, while ERα protects against vascular injury, remodeling and fibrosis, and atherosclerosis.99–101 Additionally, platelets, which are important for induction of thrombosis, express ERβ and the AR and respond to sex steroids. 102 And finally, ERα/β, PR, and AR expression has been found to shift in men and women with atherosclerosis as they age (reviewed in Ref. 6 ).
Sex hormone effects on immune cells
Our understanding of sex hormone effects on immune cells comes mainly from cell culture and animal studies of normal, healthy cells or the study of various inflammatory diseases like autoimmune diseases. Very little information exists on the effect of sex hormones on inflammation in atherosclerosis. With this paucity of data in mind, I will briefly discuss what is known generally about the effect of sex hormones on immune cells, what has been discovered about sex differences in cardiac inflammation and remodeling during myocarditis, and propose how these findings may relate to sex differences in inflammation in atherosclerosis.
Sex steroid hormone receptors like ERα, ERβ, and AR are expressed on and within immune cells that are present in atherosclerotic plaques including MCs, macrophages, DCs, T cells, and B cells. 102 Human and mouse monocytes and macrophages express ERα, ERβ, and AR.6,102 AR expression on human monocytes is higher in men compared to women.103–105 In general, estrogen has been found to have anti-inflammatory effects on macrophages. Estrogen inhibits the TLR2/TLR4 ligand lipopolysaccharide (LPS)-induced gene products like tumor necrosis factor (TNF), IL-1, and IL-6 by downregulating NFκB signaling (Table 1).106–110 Estrogen has also been found to reduce oxidative stress in healthy murine peritoneal macrophages, 110 and to skew macrophages to a M2 phenotype. 111 In contrast, Rettew et al found that testosterone decreased TLR4 expression in the RAW tumor macrophage cell line (which are male cells), 112 while ovariectomy and estrogen replacement in female C57BL/6 mice increased TLR4 expression on macrophages. 113 It is possible that the effect of sex hormones on normal healthy immune cells is not always the same as their effect during infection and disease (ie, bacterial LPS, viral infection, myocarditis). In support of this idea, we found that TLR4 expression was higher on male than female macrophages (and MCs) during innate coxsackievirus B3 (CVB3) infection and acute viral myocarditis. 114 These findings highlight some of the difficulties inherent in studying the effect of sex hormones on immune cells. Evidence that estrogen may promote an anti-atherosclerotic phenotype in macrophages comes from reports that estrogen decreases oxLDL115,116 and increases ApoE levels 117 (Table 1). Higher expression of ERα in the vasculature of premenopausal women correlates with a lower incidence of atherosclerosis, further suggesting that ERα protects against atherosclerosis.118,119
Effect of estrogen on the immune response.

Women have an increased antibody response to infections and vaccines compared to men.120,121 This is due to the ability of estrogen to activate B cells, resulting in increased levels of antibodies (and autoantibodies) (Table 1), while androgens decrease B-cell maturation reducing B-cell synthesis of antibody in humans.122–124 However, depending on the dose, estrogen can either drive Th1 and/or Th17 responses (low dose) or increase the regulatory arm of the adaptive immune response by enhancing tolerogenic DCs, IL-4-driven Th2 responses, IL-10, TGFβ, Treg, and alternatively activated M2 macrophages (high dose) (Table 1).7,122–125
Far less research has been conducted on the effect of androgens on immune cell function, but in general, androgens are thought to drive cell-mediated and Th1 responses.126–129 One complication in understanding the role of sex hormones like testosterone on immune cells is that most published studies do not interpret data in the context of sex. For example, the sex of cells used in culture experiments is seldom reported. 130 Additionally, effects prescribed to testosterone may be due to testosterone or estrogen because of aromatase conversion of testosterone to estrogen.
Sex hormone effects on myocardial inflammation
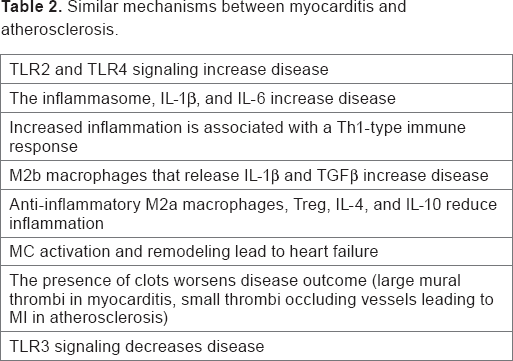
Myocarditis, or inflammation of the myocardium, shares many similarities to atherosclerosis (Table 2). The primary difference between the diseases is location; atherosclerotic inflammation is confined to the vascular wall, while myocardial inflammation is present in the heart muscle. The different locations for inflammation produce different disease outcomes (ie, dilated cardiomyopathy vs MI), but otherwise the mechanisms that promote or protect against both diseases are remarkably similar and include the following: TLR2 and TLR4 signaling increase inflammation30,35,131,132; the inflammasome and IL-1β promote disease23,30,34,53; increased inflammation is associated with a Th1-type immune response27–30,133,134; M2b macrophages that express TLR2/TLR4 and release IL-1β and TGFβ promote remodeling29,42,135; anti-inflammatory M2a macrophages, Treg and other regulatory T cells, IL-4, and IL-10 reduce inflammation65,66,114,135; MC activation promotes remodeling, thrombosis, and heart failure,38,44,51–53; and TLR3 signaling protects against disease.45,136,137 Although inflammation during atherosclerosis is initially present within the vessel wall, once an MI occurs, the myocardium is damaged and an inflammatory response ensues that is very similar to the pathogenesis of viral myocarditis.
Similar mechanisms between myocarditis and atherosclerosis.
So what about sex differences in the myocardial immune response? Men (and male mice) are more likely to develop myocarditis and progress to dilated cardiomyopathy (DCM) and heart failure than women,114,138 similar to atherosclerosis. Our understanding of sex differences in cardiac inflammation during myocarditis comes almost exclusively from animal models of disease. Huber and colleagues were the first to describe that male BALB/c mice have increased CVB3-induced myocarditis compared to females. 139 Male mice with CVB3 myocarditis develop a predominantly Th1-type adaptive immune response with more Th1-induced IgG2a anti-CVB3 antibodies compared to female mice, who develop a Th2 response and more regulatory T-cell populations.114,134,140 This Th1 vs Th2 dichotomy, as well as the severity of myocardial inflammation, has been found to be due to testosterone and estradiol, respectively.53,114,135,140–142
Males also have more MCs and macrophages than females during myocarditis, which express more TLR4 and IL-1β.53,114,135 The predominant Th1 response in male mice with CVB3 myocarditis is not due to classical IL-12/STAT4-induced IFNγ production, but TLR4-induced IL-18, a cytokine that strongly induces IFNs. 134 Male mice have more classically activated M1 macrophages in the heart during acute CVB3 myocarditis, while females have more inhibitory M2a macrophages.135,143 TLR4 mRNA expression has been found to be higher in patients with biopsy-proven myocarditis and DCM than controls.144,145 Although sex differences in TLR4 levels were not examined, ∼75% of the myocarditis and DCM patients in these studies were men. Testosterone has been found to be responsible for increasing each of these immune mediators in animal models. Additionally, cholesterol uptake in macrophages is associated with increased myocardial inflammation,146,147 similar to its role in atherosclerosis.
Evidence that estrogen protects against myocardial inflammation following MI also exists from animal models. ERα knockout mice have more ischemic damage to the myocardium compared to wild-type (WT) mice.148,149 Also, administration of an ERα-selective agonist or ERα transgenic mice were found to reduce injury and inflammation in an ischemic reperfusion injury model.101,150–152 Estrogen also protects against hypertension in females in animal models by reducing AngII levels. 153 However, hypertension increases in women after menopause when estrogen levels drop. 8 This may also be due to smaller vessel sizes in women and other physiological differences as well as changes in blood pressure related to autoimmune diseases like SLE. Overall, these data indicate that estrogen reduces myocardial inflammation while testosterone promotes it.
Sex hormone effects on cardiac remodeling
A critical step in the progression from myocarditis to DCM involves extracellular matrix (ECM) remodeling and fibrosis. 154 Scar tissue/collagen laid down in the myocardium following myocarditis is due primarily to immune cell release of profibrotic enzymes (ie, collagenases, MMPs), cytokines (ie, TGFβ1, IL-1β), and other mediators during the peak of acute inflammation. 53 The immune cells and ECM components involved in this process are virtually identical in myocarditis, atherosclerotic plaques, and post-MI cardiac damage.4,30,142 MCs and macrophages are particularly important in regulating the degradation and synthesis of ECM components that form the outer layer of the plaque leading to an MI or that produce myocardial scar tissue and DCM.30,52,155 Cytokines associated with Th2-type immune responses like IL-4, TGFβ, and IL-1β, certain M2 macrophages (ie, M2b), and MCs are central to the remodeling process. MC activation during myocarditis is associated with increased remodeling, DCM, and heart failure in males.44,53 It is important to realize that remodeling is a cycle of laying down and breaking up ECM products like collagen, so that the same process that lays down collagen to produce DCM also breaks it up to produce an MI.
Only males of mouse strains that respond to CVB3 infection with a dominant Th2 response, like BALB/c mice, develop fibrosis and DCM following acute myocarditis.44,53,136 These findings highlight the importance of Th2-driven MCs and M2 macrophages that express TLR4 and IL-1β (ie, M2b subtype) in the progression to fibrosis and DCM in males during and following acute myocarditis. This type of Th2-driven immune response appears to be disguised as a classical Th1 response in males due to elevated levels of TLR4-induced IL-18, which increases IFNγ production and Th1-type immune cells. Interestingly, macrophage-derived IL-18 has recently been found to increase fibrinogen deposition during vascular remodeling in rats, 156 suggesting that the inflammasome is particularly important for plaque remodeling in men with atherosclerosis.
Several studies report that patients with myocarditis or DCM have a greater induction of ECM proteins and/or fibrosis in the heart compared to women, including increased collagen and MMP production.92,157,158 In myocarditis patients, men are 2 times more likely to present with myocardial fibrosis then women by cardiac magnetic resonance imaging (MRI). 157 Various animal models of heart disease have shown that testosterone is responsible for adverse cardiac remodeling in males following MI 159 and that estrogen signaling via ERα, in particular, prevents cardiac myocyte hypertrophy and fibrosis in females by blocking TGFβ and collagen synthesis.99–101 Serpin A3n, also known as α1-anti-chymotrypsin, is an important remodeling enzyme released from MCs, which was found to be upregulated in 11 separate microarray studies of acute DCM patients as well as in male mice with acute CVB3 myocarditis.53,160 Serpin A3n was found to be increased in the heart of male mice by testosterone and IL-1β and to induce cardiac fibrosis by altering MMPs during acute CVB3 myocarditis. 53 Overall, these studies suggest that elevated testosterone levels in men increase cardiac inflammation and fibrosis, leading to DCM and heart failure. Although few studies have specifically examined sex differences in remodeling during atherosclerosis, a similar role for sex hormones in the remodeling process would be expected for atherosclerosis.
Sex hormone effects on inflammation with aging and menopause
Age-related changes in immune function include an impaired ability to respond to new antigens, unsustained memory responses, chronic low-grade inflammation, and an increasing propensity for autoimmune responses due to antibody and autoantibody levels that increase with age.161–163 Few studies exist examining sex hormone effects on the immune response with aging in atherosclerosis, except for studies on brain inflammation, aging, and stroke. 164 Hormone replacement therapy (lifting estrogen levels to a relatively higher dose) decreases antibody levels in menopausal women, 165 suggesting that these changes are age-related rather than due to estro-gen levels alone. Menopause is defined as the final menstrual period without another menstrual period for 12 months.166–168 The mean age when menopause occurs in Western cultures is between 49 and 52 years.166,167 Decreases in estrogen (ie, estradiol) occur only in the last six months before menopause and thereafter. 168 In contrast, testosterone gradually decreases with age in both sexes.169–171 After menopause, the primary endogenous source of estrogen is estrone, which is synthesized in adipocytes via aromatase. 172
Menopause is also associated with decreases in CD4+ and CD8+ T cells and natural killer cell activity.172–174 Additionally, aging and menopause have been associated with an increase in the cytokines IL-1, IL-6 (IL-1β increases IL-6 levels), IL-18, TNF, and C-reactive protein (CRP) levels, which are biomarkers of an inflammatory response and associated with increased risk for heart failure in atherosclerosis patients.6,156,175,176 Importantly, these cytokines are derived mainly from innate immune cells like macrophages and MCs at sites of tissue inflammation. That lower estrogen levels during menopause increase these cytokines is consistent with the known ability of higher doses of estrogen to downregulate NFκB, Th1 responses, IL-1, and IL-6 via ERα in various human and murine cell types.107,109,177–179 Thus, during menopause the protective role of estrogen in decreasing innate immune cell activation is lost, allowing increased proinflammatory cytokine levels, while, at the same time, autoantibody levels and ICs continue to rise due to continued low levels of estrogen. In contrast, in most men, androgen levels that promote innate immune cell activation and proinflammatory cytokines decline past age 50 without the increase in autoantibodies found in women because of estrogen. This may explain, at least in part, why atherosclerosis and hypertension increase in women following menopause.5,6
Implications for sex differences in inflammation during atherosclerosis
The similarity between immune mechanisms involved in the pathogenesis of myocarditis and atherosclerosis suggests that sex differences in the inflammatory response and cardiac remodeling may also be similar. However, mechanistic studies examining sex differences in inflammation during atherosclerosis have, for the most part, not yet been conducted. A number of key questions need to be addressed. Do sera biomarkers like TNF and IL-6 predict poor outcome in male and female atherosclerosis patients? Or, are they better predictors in males due to the inhibition of these cytokines by estrogen? Do TLR4/inflammasome/IL-1β levels predict MI in males better than females because testosterone increases this signaling pathway? Is the correlation between autoantibodies to oxLDL and CAD more prevalent in females where estrogen increases B-cell and antibody/autoantibody responses? If so, could this partially explain why women are at an increased risk to develop CAD when they have one or more autoimmune diseases? Why is hypertension higher in women with CAD than men after menopause, and does hypertension relate directly to cardiac inflammation? Does AngII activation promote hypertension and arrhythmias more in men because estrogen reduces its expression in women? These are just a few of the questions that need to be answered to gain a better understanding of sex differences in the pathogenesis of atherosclerosis and risk factors for MI.
If sex differences in inflammation found in myocarditis apply to atherosclerosis, I hypothesize that MCs and M2b TLR4+ macrophages increase inflammation and foam cell accumulation in the atherosclerotic plaques of men where they promote remodeling resulting in a thrombotic MI (Fig. 2). In contrast, a primary driver of MIs in women could be the result of deposition of autoantibodies against oxLDL and other autoantibodies in the form of ICs (Fig. 2). The presence of ICs coupled with the smaller size of vessels could increase blood pressure in women and promote local inflammation and shearing of the vessel wall to generate an MI. This process would be accelerated after menopause in women as cardioprotective estrogen levels drop but who have elevated autoantibody levels due to the presence of one or more autoimmune diseases.

Atherosclerotic inflammation leading to MI in men vs women. Testosterone increases the number and activation of mast cells (MCs) and macrophages (Mac), leading to the development of TLR2+/TLR4+/M2b macrophages and foam cells within atherosclerotic plaques (upraised area of inflammation). Cytokines, enzymes, and other mediators like MMPs released from MCs and Mac allow remodeling of the vessel wall, resulting in thrombus formation and MI in men. In contrast, estrogen promotes more antibodies and autoantibodies (AutoAbs) against oxLDL, for example. Deposition of ICs on narrow vessel walls activates complement and leads to shear stress from higher blood pressure in females, thrombus formation, and MI in women.
Conclusions
We currently do not understand how the immune response in women differs from men with atherosclerosis because most clinical studies contain a majority of men, which skews our understanding of the factors important for men, and data are typically not analyzed according to sex (controlling for sex does not adequately reveal sex differences). Additionally, researchers studying atherosclerosis using animal models have not reported sex differences in disease. In order to improve diagnosis and treatment of CAD, the research community needs to understand how the immune response is similar and how it differs in men and women with atherosclerosis.
Author Contributions
Wrote the manuscript: DF. Developed structure and arguments for the paper: DF. Made critical revisions and approved final version: DF. The author reviewed and approved of the final manuscript.