
Abstract
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is a genetic form of cardiomyopathy (CM) usually transmitted with an autosomal dominant pattern. It primary affects the right ventricle (RV), but may involve the left ventricle (LV) and culminate in biventricular heart failure (HF), life threatening ventricular arrhythmias and sudden cardiac death (SCD). It accounts for ll%-22% of cases of SCD in the young athlete population. Pathologically is characterized by myocardial atrophy, fibrofatty replacement and chamber dilation.
Diagnosis is often difficult due to the nonspecific nature of the disease and the broad spectrum of phenotypic variations. Therefore consensus diagnostic criteria have been developed and combined electrocardiography, echocardiography, cardiac magnetic resonance imaging (CMRI) and myocardial biopsy. Early detection, family screening and risk stratification are the cornerstones in the diagnostic evaluation. Implantable cardioverter-defibrillator (ICD) implantation, ablative procedures and heart transplantation are currently the main therapeutic options.
Keywords
Introduction
Arrhythmogenic right ventricular cardiomyopathy/ dysplasia (ARVC/D) is a genetic form of cardiomyopathy that primarily affects the right ventricle (RV). It may also involve the left ventricle (LV) and culminate in life-threatening ventricular arrhythmias, prompting sudden cardiac death (SCD) and/or biventricular heart failure.1–5 ARVC/D was first described by the Pope's physician, Giovanni Maria Lancisi, in his book entitled De Motu Cordis et Aneurysmatibus, published in 1736. 6 He reported that there was a family who had experienced pathologic RV, heart failure and SCD in four generations. The first comprehensive clinical description of the disease was reported by Marcus et al in 1982, when he reported 24 adult cases with ventricular tachyarrhythmias with left bundle branch morphology. 7 Nevertheless, it was not until 1984 that the electrocardiographic features of the disease were first described, including the epsilon wave. 8
ARVC/D is characterized pathologically by myocardial atrophy, fibrofatty replacement, fibrosis and ultimate thinning of the wall with chamber dilation and aneurysm formation. 9 These changes consequently produce electrical instability precipitating ventricular tachycardia (VT) and SCD.10–20
Epidemiology
The estimated prevalence of ARVC/D in the general population is approximately 1:5000, affecting men more frequently than women with a ratio of 3:1.21,22 ARVC/D accounts for 11%-22% of cases of SCD in the young athlete patient population,23–25 accounting for approximately 22% of cases in athletes in northern Italy 26 and about 17% of SCD in young people in the United States. 27
It has been difficult to determine the incidence of ARVC/D due to the different clinical manifestations of the disease. These manifestations vary greatly, especially in different ethnic groups. This is probably secondary to its genetic heterogeneity and variable phenotypic expression, along with a diverse disease progression, which make diagnosis difficult, and in turn, decreases the real incidence evaluation of this entity.
These ethnic variations are likely caused by different modes of inheritance and involved genes. For instance, in Naxos Island, Greece, the mode of inheritance of ARVC/D in association with palmoplantar keratosis, also known as Naxos disease, was demonstrated to be autosomal recessive, 28 whereas the mode of inheritance in most of ARVC/D is autosomal dominant. Other studies suggest that Chinese patients may have a lower familial incidence of premature SCD among ARVC/D patients compared to Western population, although there is no genetic data on this population. 29
Disease Genetics and Pathogenesis
ARVC/D was initially believed to be secondary to a developmental defect of the RV myocardium, leading to the original designation of “dysplasia”, similar to Uhl's anomaly. 30 Nonetheless, this process differs from ARVC/D based on the fact that Uhl's anomaly has not been documented to have a genetic basis, and it is not recognized as a desmosomal disease. In addition, myocardial atrophy is the consequence of cell death after birth and its progressive postnatal development has been definitively assessed (See Table 1).26,31,32 This concept has evolved over the last 30 years and based on its clinical characteristics, pathophysiology, post-natal development and genetic background, its inclusion in the World Health Organization (WHO) classification of cardiomyopathies was finally achieved.33,34
Differences between Uhl's anomaly and ARVC/D.
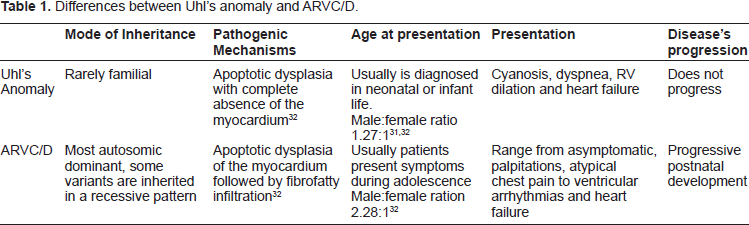
Several studies have been performed to determine the etiology and pathogenesis of ARVC/D. However, there still is conflicting evidence. Different causes have been suggested such as congenital defects, genetics and acquired factors. The hypothesis of a congenital cause leads to the adoption of the term dysplasia due to its association with Uhl's anomaly, as explained earlier, but ARVC/D has been differentiated and its natural history through adult life has been well documented.3,30 This hypothesis has evolved into the idea of genetically determined cardiomyopathy, which has been retained on genetic grounds.33,34 In approximately 30%-50% of cases it is transmitted with an autosomal dominant pattern of inheritance, with incomplete penetrance and variable expression.35–37
Acquired factors have also been suggested as the cause of ARVC/D. The strongest association has been made with viral myocarditis inducing arrhythmogenic cardiomyopathy. On histopathology, a lymphocytic infiltrate with disappearance of myocytes and fibrofatty replacement is often found, which can also be seen in viral myocarditis.38,39 It is possible that the wide variation in presentation of ARVC/D patients could be explained by its genetic heterogeneity and modifying factors such as exercise and/or viral myocarditis.23,40,41
The genetic hypothesis has been thoroughly studied. The first chromosomal locus identified was published by Rampazzo 44 et al in 1994 in Italy. Linkage analysis supported the evidence for genetic heterogeneity of several ARVC/D loci on chromosomes 1, 2, 3, 6, 10, 12 and 14.42–45 Similarly, he reported the Desmoplakin gene (DSP), the first desmosomal protein gene to be associated with a major form of the disease, with autosomal dominant inheritance, also called ARVC/D type 8 46 (See Table 2).46–51
Different types of ARVD/C and its genetics.

In addition, the gene for Naxos disease was mapped on chromosome 17 (locus 17q21), by McKoy et al.28,43 This is the first disease-causing gene, also named junction plakogoblin (JUP) gene (autosomal recessive variant of ARVC/D) characterized by palmoplanar keratosis, woolly hair and ARVC/D. The association between skin and heart manifestations could be explained by the finding that epidermal cells in the soles, palms and myocyte share a similar mechanical junctional apparatus (fascia adherens and desmosome) responsible for cell-to-cell adhesion, and are exposed to the high frictional stress. The JUP gene encodes the desmosomal protein, plakoglobin, a major cytoplasmic protein that is the only known constituent common to submembranous plaques of both desmosomes and intermediate junctions responsible for cell adhesion. This suggests that ARVC/D is a cell-to-cell junction disease, leading detachment of the myocytes and fibrosis, particularly under conditions of mechanical stress.21,28,43,46,52
A recessive mutation of DSP has been reported and associated with Carvajal syndrome, another cardiocutaneous disease. 53 Plakophilin-2 (PKP2) is the most frequent targeted protein with more than 25 different mutations identified in the gene encoding it. 52 Thus, ARVC/D was found to be mainly a disease of the cardiomyocyte junction. 54 (See Table 3).
Cardiocutaneous disorders associated with ARVC/D.

Furthermore, extradesmosomal genes, unrelated to the cell adhesion complex, have been implicated as autosomal dominant forms of ARVC/D, such as (1) the cardiac ryanodine-2 receptor gene, responsible for the release of calcium from the sarcoplasmic reticulum; (2) the transforming growth factor-β3 gene (TGFβ3), implicated in the regulation of the production of extracellular matrix and expression of genes encoding desmosomal proteins and the TMEM43 genes.55–57
Clinical Presentation
ARVC/D is a devastating disease given the fact that the first symptom is often SCD, which makes early detection and a family screening test the cornerstone in the diagnostic evaluation. In a study of 100 patients with ARVC/D, Dalal et al reported an incidence of SCD of 23% in the United States. Other common presentations were palpitations (27%) and syncope (26%). 23 Early intervention with an Implantable Cardioverter-Defibrillator (ICD) decreases the risk of SCD, but ARVC/D is a progressive disease, which can progress to refractory/recalcitrant VT or a ventricular fibrillation (VF) storm despite ICD therapy.
Other studies have suggested that up to up to 67% of individuals with ARVC/D present with palpitations, 32% present with syncope, 27% with atypical chest pain, 6% with RV failure and 6% may remain asymptomatic. (See Fig. 1). 58 The most common arrhythmia is sustained or nonsustained monomorphic VT that originates in the RV and therefore has a left bundle branch block (LBBB) morphology. Alternatively, in some cases where the LV is involved, VT could present with a right bundle branch block (RBBB) morphology. These symptoms are usually exercise-related, mainly due to an activation of the sympathetic nervous system, which may lead to premature beats and re-entrant arrhythmogenic circuits. In a pathological myocardial substrate, this can perpetuate the incidence of life-threatening arrhythmias.
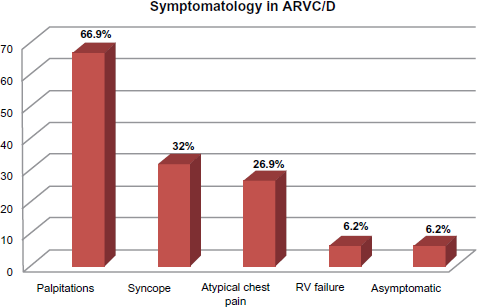
Presenting symptoms and frequencies in patients with ARVC/D. 58
The advanced stage of ARVC/D is characterized by signs and symptoms of RV or LV failure depending on which ventricle is more affected (in the classic form, it is generally the RV more affected than the LV), and finally frank biventricular congestive heart failure, which may be difficult to distinguish clinically from dilated cardiomyopathy. (See Fig. 2). 58

Incidences of ventricular involvement in ARVC/D. Classic form are mostly RV involvement, LV or Biventricular involvement. 58
Clinicopathologic Manifestations
The genotypic abnormalities explained above translate into phenotypic presentations with variable penetration, leading to the anatomoclinic profile of ARVC/D.
The histological examination of the myocardium reveals a typical fibrofatty infiltration with areas of surviving myocytes and inflammation progressing from epicardium to endocardium. This infiltration may be diffuse or regional; the latter is typically located at the angles of the “triangle of dysplasia” at the infero-apical and infundibular walls. This occurs earlier in the RV, compared to the left side of the heart, but LV involvement may also sporadically occur in early stages of the disease.8,33,59 Parietal thinning as well as endocardial thickening is found in areas of transmural infiltration and aneurismal dilation providing a substrate for life-threatening arrhythmias.
Biventricular involvement with LV fibrofatty replacement and involvement have been found as much as in 70% of the cases of ARVC/D. It is usually age dependent, associated with more severe cardiomegaly, arrhythmogenic events, inflammatory infiltrates and heart failure. ARVC/D can no longer be regarded as isolated disease of the RV. 60 Accordingly, several authors have opted for naming this condition “Arrhythmogenic Cardiomyopathy” but the term “Right Ventricular Arrhythmogenic Cardiomyopathy” is still widely used.
The natural history of ARVC/D is highly variable, with a wide spectrum of clinical presentations. Four patterns have been proposed:24,33 (1) Concealed form, minor arrhythmias, which usually go unnoticeable. The diagnosis is usually made during family screening. 61 (2) Overt Electrical Heart disorder, the most typical presentation, which usually occurs in young patients presenting with severe and symptomatic ventricular arrhythmias and SCD. 62 (3) RV failure, in which the myocardial replacement in the RV leads to RV dysfunction with pump failure. (4) Biventricular failure characterized by progressing dilation of RV and LV; this usually develops late in the natural history of the disease.3,60 Other conditions have also been associated such as valvulopathies (ie, mitral valve prolapse 63 and tricuspid valve prolapse) and patent foramen ovale.64,65 The mechanism of this association is still not known. We have postulated two different mechanisms: (1) raised left atrial pressure, as seen in patients with heart failure as they can dilate the foramen ovale leading to left-to-right shunt, while RV failure and tricuspid regurgitation can lead to rise in right atrial pressure and cause right-to-left shunt. (2) Apoptotic dysplasia, genetically determined by chromosome abnormalities, leading to loss of cell-to-cell adhesion and consequently patent foramen ovale and/or valvulopathies.
It is important to emphasize that fibrofatty infiltration is possibly the least reliable criterion for the diagnosis of ARVC/D, 66 as fat may also be part of scarring from ischemia, inflammation and myocyte vacuolization, as seen in other cardiomyopathies. Therefore, its histopathological diagnosis should be done in conjunction with other features: fibrosis, tissue deposition, myocytes and inflammatory infiltration.
Diagnoses
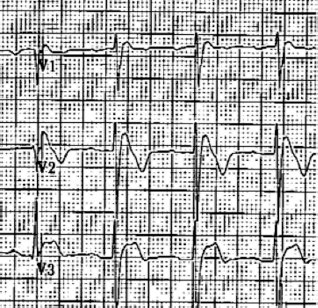
ARVC/D should be suspected in a young patient with palpitations, syncope or aborted SCD. VT with LBBB morphology is the classic presentation, but as mentioned above, VT with RBBB may be present if the LV is involved. Other electrocardiographic abnormalities such as inverted T waves in right precordial leads (V1-V3) and frequent premature ventricular complex (PVCs), even in asymptomatic patients, should arouse the suspicion for this cardiomyopathy. 67
Clinical diagnosis of ARVC/D is often difficult because of the non-specific nature of the disease and the broad spectrum of phenotypic variations. There is no definitive diagnostic standard. Endomyocardial biopsy (EMB) of the RV is definitive (gold standard) when positive but it often yields a false-negative result and in most patients, assessment of transmural myocardium is not possible based on the fact that the disease spreads from epicardium to endocardium. Therefore, it is not practical in the clinical setting.68,69 The sensitivity of EMB is approximately 67% because of the regional fibroadipose infiltration. 70 Consequently, the best approach in making a diagnosis of ARVC/D is by combining different diagnostic tests.
Several investigations contribute to the diagnosis of ARVC/D. Consensus diagnostic criteria have been developed, which include RV biopsy, noninvasive electrocardiography, family history and imaging evaluation with echocardiography, cardiovascular magnetic resonance imaging (CMR) and angiography.71,72 The advent of the molecular genetics era has made a tremendous diagnostic impact and contributed new insights to the understanding of its pathophysiology. 21
In the last decade, enormous advances have been made, which include two research grants in Europe and USA achieved by Basso et al and Marcus et al respectively.73,74 These have greatly contributed to the disease characterization, diagnosis, pathophysiology and therapeutic options.
The 1994 Task Force criteria were based upon clinical experience, family history, and structural, functional and electrocardiographic abnormalities in patients with severe disease. 35 Thus, these criteria have high specificity but lack sensitivity, increasing the risk of a false negative in the early stages of ARVC/D. A modification of these original criteria was made in order to include first degree relatives on early stage of the disease or incomplete expression. This increased the sensitivity, but not surprisingly also increased the risk of false positives cases.75,76 These diagnostic criteria were revised in 2010 and now incorporate the advances in both technology and genetics (See Table 4). 72
2010 revised task force criteria.

Modified from: Marcus FI, McKenna WJ, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. Apr 6, 2010;121(3):1533-41.
The diagnosis is based on the presence of major and minor criteria, which are classified into six categories. The diagnosis of ARVC/D is made in the presence of two major or one major plus two minor criteria or four minor criteria taken from different groups. (See Fig. 3). 72

Diagnosis of ARVC/D: major and minor criteria. 72
Diagnostic Modalities
An epsilon wave (depolarization abnormality) may be found in 30%-33% of patients with ARVC/D 58 and it is described as a distinct wave at the end of QRS complex (see Fig. 4) probably secondary to slowed intraventricular conduction.
Epsilon wave: low amplitude positive deflection at the end of the QRS complex, usually seen in V1 and V2. If the epsilon wave is of large magnitude a reciprocal epsilon wave may be seen in V5 or V6, this may suggest that large part of the RV is depolarizing late.
This is also a major criterion in the 2010 revised Task Force criteria (see Table 4, IV). 72 Ventricular ectopy with LBBB morphology is also highly suggestive of ARVC/D. The origin of the ectopic beats is usually from one of the three regions of fatty degeneration in the “triangle of dysplasia”. (See Table 5).23,58,79,80
Incidence of ECG findings in ARVC/D. ECG changes in ARVD/C.

Diagnostic accuracy of noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging.82. Delayed enhancement magnetic resonance imaging. 4-chamber and short axis views of DE-CMR showing delayed enhancement of the free right ventricular wall.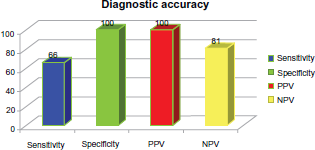
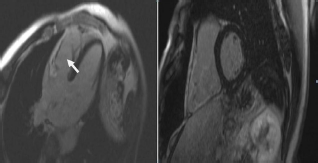

The imaging is a Haematoxylin and eosin stained section and shows the typical fibrofatty infiltration of the ventricles in ARVC/D.
Other tests
Despite all these different diagnostic modalities, no test is perfect. Diagnosis is difficult and requires the combination of all tests, if possible. Recent advances in technology promise to make DE MRI the imaging modality of choice to assess myocardial diseases. 82 Nevertheless, this modality is not included in the current modified Task Force criteria (See Fig. 6). 82
Differential Diagnosis
Right ventricular outflow tract tachycardia (RVOT) is one of the main differential diagnoses of ARVC/D. The presentation and electrocardiographic characteristics of VT could be very similar in these two (VT with LBBB but with an inferior axis). However, RVOT tachycardia is considered to be a primary electrical disease in the absence of structural heart disease, unlike ARVC/D. Epidemiologically, RVOT tachycardia is more common than ARVC/D.
Other conditions that should be differentiated from ARVC/D are: cardiac sarcoidosis, 83 pre-excited AV re-entry tachycardia, Brugada syndrome (See Fig. 9), pulmonary hypertension, tricuspid valvulopathy and other congenital heart disease such as Uhl's anomaly (See Table 1), and repaired Tetralogy of Fallot, among others. (See Table 6).
Brugada syndrome: characterized by a dynamic ST-segment elevation (accentuated J wave) in leads V1 to V3 of the ECG followed by negative T wave.
Differential diagnosis of ARVC/D.

Treatment
As discussed previously, patients with ARVC/D may remain asymptomatic or may present with a wide variety of symptoms. Therefore, the therapeutic strategy has to be individualized, based on clinical presentation, risk stratification and patient/physician preference.22,60,83–86 The main goal of therapy is to prevent serious events, which requires identifying high-risk patients for malignant arrhythmias and SCD.87–89
Although several studies have evaluated the benefit of pharmacological and non-pharmacological therapies in patients with ARVC/D,87–91 large prospective randomized trials are not available. Consequently, the current therapeutic recommendations for this entity have been developed from observational studies and case series, or have been adopted from other cardiomyopathies,92,93 where solid evidence supported by clinical trials is available and clear guidelines are defined.87,89
Physical activity
Exercise has been related to an increased incidence of ventricular conduction disorders, SCD, worsening symptoms and progression of the fibrofatty atrophy,22,78,94–96 particularly in young patients and competitive athletes.86,97 In 1996 Leclercq et al 95 described the association between progressive sympathetic stimulation present during physical activity and the onset of monomorphic sustained VT. The initiation of the VT was associated with the typical catecholamine surge seen on exercise in the setting of the presence of arrhythmogenic substrate characteristic of these patients.22,95 For that reason, high risk patients with suspected or confirmed ARVC/D diagnosis should avoid vigorous physical activity including competitive sports, regular training and strenuous exertion involving abrupt physical effort, as well as any recreational activity associated with symptoms (Level of Recommendation: IB).96–98
Pharmacological therapy
Antiarrhythmic medications have been used for symptomatic control in patients who are not candidates for an implantable cardioverter-defibrillator (ICD) or as an adjunct therapy to reduce frequent ICD discharges due to recurrent VT.89,99 Antiarrhythmic medications have been mostly recommended in patients with severe LV dysfunction and severe symptoms of ventricular arrhythmias. Beta-adrenergic blocking agents have shown effectiveness in reducing adrenergically-stimulated arrhythmias.22,85,100,101 Nevertheless, the evidence available has been derived from observational studies, which have shown conflicting results.
The combination of beta-blockers and amiodarone 100 have had a beneficial effect in suppression of non-sustained VT, reduction in the frequency of sustained ventricular arrhythmias, 91 and reduction of VT rate preventing syncope and favoring antitachy-cardia pacing termination rather than shock therapy. Furthermore, they favor the suppression of other arrhythmias, especially supraventricular tachycardia (SVT) and atrial fibrillation which may cause symptoms or interfere with ICD function, resulting in inappropriate discharges. 91
Sotalol and amiodarone have been proposed as effective treatment of sustained VT or ventricular fibrillation (VF) as adjunctive therapy to ICD or in patients with ARVC/D that are not candidates for ICD implantation (Level of Recommendation: IIaC).89,91,99 In 1992, Wichter et al 91 published that sotalol was the most effective medication in the treatment of both inducible and non-inducible VT with efficacy rates of 68.4 and 82.8% respectively, in patients with ARVC/D; patients who did not respond to sotalol were non-responders to other antiarrhythmic therapies including amiodarone. Class I antiarrhythmic drugs were effective in only a minority of patients (14.8% and 18.5%), and beta-blockers did not show effectiveness in patients with inducible VT and were only partially effective in patients with non-inducible VT (28%). Amiodarone was not superior to sotalol, being effective in 15.4 and 25% of the respective groups, and verapamil was more effective in non-inducible VT group (0% vs. 50%).
In contrast to the results published by Wichter, in 2009 Marcus et al 102 reported that neither beta-blockers nor sotalol seemed to be protective against clinically relevant ventricular arrhythmias. Moreover, the sotalol group was found to have a potential increased risk for significant ventricular arrhythmias or ICD therapy. However, is important to recognize that high risk patients for major arrhythmias were most likely to be on sotalol. On the other hand, amiodarone, although only received by a relatively small number of patients, had the greatest efficacy in preventing ventricular arrhythmias. This study was done in 95 patients with ICD implantation selected from the North America ARVC Registry. These conflicting results need to be further addressed in future trials.
Non Pharmacologic Treatment
Implantable cardioverter-defibrillator (ICD)
The indications of ICD for primary prevention of SCD in ARVC/D patients have not been well established.87,89 It represents prophylactic implantation based on a clinical profile with 1 or more identifiable risk factors for SCD. There is a consensus that high-risk patients should be considered as ICD placement candidates. Consequently, patients with episodes of sustained VT or VF (Level of Recommendation: IB), unexplained syncope, non-sustained VT on noninvasive monitoring, familial history of sudden death, extensive disease including those with LV involvement and good functional status (Level of Recommendation: IIaC)87,89,103 are potential candidates for ICD implantation even in the absence of ventricular arrhythmias.104,105 Additionally, patients with genotypes of ARVC/D associated with a high risk for SCD (eg, ARVC/D 5) should be considered as possible candidates for ICD therapy.87,104 It is currently recommended that asymptomatic patients have to be managed in a case by case basis.
In 2005, Hodgkinson et al 104 identified 11 families from Newfoundland, Canada who were affected by autosomal-dominant ARVC/D5, which is related to mutation at chromosome 3-3p25 and is associated with early SCD, especially in men. 48 high risk patients were followed after ICD implantation (35 patients) (73%, 17 males and 18 females) for primary prevention and 13 patients (27%, males) for secondary prevention. Likewise, 58 matched high-risk patients without ICD were selected as a control group (32 males and 22 females). The authors reported a five-year mortality rate of 0% for males after ICD implantation compared to 28% in controls. In this study, homogeneous ARVC/D5 patients were studied in order to evaluate the real impact on mortality of ICD implantation. However, its clinical applicability may be reduced to patients with this specific genetic defect and may not be applied in practice to all patients with ARVC/D.
There are not specific prospective randomized trials for secondary prevention of SCD in an ARVC/D population, or comparing medical therapy versus ICD. Some observational studies have shown adequate ICD use for malignant ventricular arrhythmias/SCD in patients with ARVC/D.93,104,106–110 Nevertheless, there are well-defined recommendations for ICD placement in patients who survived to a cardiac arrest due to VF, or after facing a sustained VT with hemodynamic compromise with no evidence of reversible causes (Level of Recommendation: IA).87,88,93,110 For secondary prevention of SCD, ICD placement has improved survival compared with pharmacological options, regardless of the underlying myocardial disease.87,89,111–114
Corrado et al described an observational study focused in the impact of ICD therapy on SCD prevention in an ARVC/D population. 93 They followed 132 patients after successful implantation due to cardiac arrest, sustained VT with and without hemodynamic compromise, unexplained syncope and family history of SCD due to ARVC/D. During a mean follow-up of 3.3 years, they observed that 48% of the subjects received appropriate ICD intervention (rate of 15% per year) for episodes of VTs with a similar incidence of appropriate termination in patients classified by clinical presentation. The total patient survival rate of 96% in patients with ICD compared with a 72% of the projected SCD-free survival rates without the device in 3.3 years (P < 0.001). Significant predictors of malignant ventricular arrhythmias, such as history of cardiac arrest or VT with hemodynamic compromise (10% incidence per year despite antiarrhythmic drug), unexplained syncope (8% per year), younger age and left ventricular involvement were identified.
Bhonsale et al 115 recently published important predictors of appropriate ICD intervention in patients with ARVC/D who received ICD implantation for primary prevention of SCD, which include: proband status, the presence of NSVT, inducibility at EPS and a Holter PVC count > 1,000/24 hours, but only the presence of NSVT and inducibility after EPS remained significant predictors of appropriate ICD interventions on multivariable analysis. This is an important step in understanding and identifying predictor of appropriate therapy in these patients and developing risk stratification scheme in ARVC/D patients. 115
Complications from ICD implantation
Although there is clear evidence of the benefit of ICD implantation as a secondary prevention mechanism for SCD, the devices are not free of short- and long-term complications. The variation of the myocardium structure with fibrofatty infiltration and RV endomyocardial scarring can produce difficulties in ICD lead placement, leading to inadequate sensing and procedural complications like myocardial injury or aneurysmal rupture. 22 Patients with ARVC/D who required ICD placements are usually younger compared to patients with ICD due to other diseases. 93 This situation leads to an increased lifetime risk of device-related complications, associated with implantation, leads, pulse generator and functionality,110,116,117 as well as deleterious psychosocial effects and detriment of quality of life.118,119 Finally, the current guidelines discourage the placement of defibrillators in patients with incessant VT, VF or with significant psychiatric illnesses that may be aggravated by device implantation or that may preclude systematic follow-up. (Level of Recommendation: IIIC). 87
ICD programming
The prevention of inappropriate ICD shocks in these patients could be increased by programming certain variables which include: faster ventricular-tachycardia/ventricular-fibrillation (VT/VF) thresholds and longer detection durations, activating enhanced detection criteria up to 200 bpm and deactivating sustained rate durations, activating algorithms to detect oversensing or lead problems and applying remote monitoring. Recently the Multicenter Automatic Defibrillator Implantation Trial-Reduce Inappropriate Therapy (MADIT-RIT) was published which proves that a high-rate therapy (delivered at a heart rate of > 200 bpm) and delayed ICD therapy, as compared with conventional device programming, were associated with reductions in a first occurrence of inappropriate therapy and reductions in all causes of mortality. 120 These findings may change the current practices with two new programming approaches, which minimize the overall shock burden.121–123
Radiofrequency ablation
Radiofrequency ablation has been reserved for patients with recurrent ventricular arrhythmias despite treatment with antiarrhythmic drugs.124–126
Ablation is considered a complementary therapy to ICD (Level of Recommendation: IlaC), useful for management of symptoms but may not be sufficient to prevent SCD.87,89 There is a high rate of recurrence after endocardial ablation due to the characteristic generalized, patchy involvement of the myocardium and progressive nature of this disease.124,125 It is worth pointing out that with newer current approaches for catheter ablation, by combining endocardial and epicardial ablation we can decrease the rate of recurrent VT. Multiple recent studies suggest that simultaneous epicardial and endocardial approaches for VT mapping and ablation are feasible and might even result in elimination of recurrent VT. This could be explained by the preferential epicardial infiltration of the disease. 127
Surgical treatment
Surgical isolation of the RV free wall prevents the propagation of malignant arrhythmias from the RV to the LV; by performing this surgical procedure there would be less substrate for the electrical disturbance. This therapeutic approach was once an option for patients with VT refractory to antiarrhythmic medications.128–130 However, patients were at risk of postoperative RV failure. Over the last decades, this procedure has been replaced by ICD placement, which has achieved widespread acceptance as a preventive treatment for SCD.
In patients with late complications of the disease, who develop heart failure symptoms or life threatening and untreatable VT, heart transplantation could be an option with good short and long term survival. Heart transplantation is essentially the final therapeutic option for these patients. 131
Other Recommendations
Additionally, patients with signs of ventricular failure should be given optimal therapy for heart failure and anticoagulation should be considered in patients with atrial fibrillation, significant ventricular dilation or ventricular aneurysm. 60
Screening for asymptomatic family members
Once the diagnosis of ARVC/D has been confirmed, a systematic evaluation of the family members favors an early diagnosis of possible affected subjects.
Nava et al evaluated 365 subjects from 37 Italian families affected by ARVC/D. They found that 41% (151 subjects) of the family members were affected by the disease, with clinical manifestations present during the adolescence and young adulthood. 78 They also observed a broad clinical spectrum of manifestations with incomplete expression and progressive natural history of the disease, recommending a long-term follow up of relatives. Family members who remain asymptomatic in the early “concealed” phase could still be at risk of disease progression and/or adverse cardiovascular events. 72
In 2010, Marcus et al proposed a modification of the 1994 Task Force Criteria in order to improve the diagnostic sensitivity for early and familiar involvement, 72 incorporating evidence available from genetics and technologic advances, including improvement of echocardiographic techniques and CMR. As part of the evaluation, an ECG, 24 hour Holter monitoring, echocardiogram and, if possible, a signal averaged ECG should be obtained on all first-degree relatives of patients diagnosed with ARVC/D. Family involvement is considered if there is evidence of ECG changes including T wave inversions in V1-V3 (individuals older than 14 years in the absence of RBBB), late potentials on SAECG, episodes of VT with RV origin pattern or more than 200 premature ventricular contractions in 24 hours, as well as the RV structural or functional abnormalities previously described.72,86
Quarta et al evaluated the roll of mutation analysis on familial evaluation with ARVC/D. 132 The study enrolled 210 first-degree relatives and 49 living probands from 100 affected families. Data obtained from clinical assessment and screening of 5 desmosomal genes associated with the disease showed clinical expression in 41.9% of relatives, at least 1 mutation in 56 families and multiple desmosomal variants in 8 families. Multiple mutations were more common in probands than in relatives, and they were associated with a significant increase in risk of disease expression (ie, 5-fold in affected family members). On the other hand, the prevalence of the disease in affected first-degree relatives was similar after comparing probands with positive and negative mutation screening (28.9% vs. 20%, respectively; P = 0.57), suggesting possible non-identified genetic abnormalities. 132
Although genetic analysis in not considered a contributor to risk stratification, it could be useful in families with history of SCD. Identification of pathogenic mutations in affected patients with cascade screening of relatives may offer an alternative cardiovascular evaluation of family members. Therefore, genetic counseling may be provided, assessing hereditary risks to next generations.72,89,132
Conclusion
ARVC/D is a progressive disease with life-threatening complications, which constitute a clinical diagnostic challenge for physicians, given the different genotypic and phenotypic variations and the wide ranges of clinical manifestations. The main challenge is to improve the risk stratification for better identification of high risk patients of SCD and heart failure, who most benefit from early intervention with lifestyle changes, restriction of physical activity, antiarrhythmic drugs, ICD placement, new ablation approaches with simultaneous endocardial and epicardial ablation and, if necessary, heart transplantation. These interventions are available and life saving, with the potential to change the natural history of the disease by offering a good quality and better life expectancy.
Author Contributions
Conceived and designed the review article: JR. Analyzed the data: EM, CM. Wrote the first draft of the manuscript: EM, CM. Contributed to the writing of the manuscript: RL. Agree with manuscript results and conclusions: JR. Jointly developed the structure and arguments for the paper: EM. Made critical revisions and approved final version: JR, EM, CM. All authors reviewed and approved of the final manuscript
Funding
Author(s) disclose no funding sources.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgement
We would like to thank Dr. Sherri White of the Albert Einstein College of Medicine/Jacobi Medical Center, Bronx, New York for providing histopathological images.