
Abstract
Philadelphia positive malignant disorders are a clinically divergent group of leukemias. These include chronic myeloid leukemia (CML) and de novo acute Philadelphia positive (Ph(+)) leukemia of both myeloid, and lymphoid origin. Recent whole genome screening of Ph(+)ALL in both children and adults identified an almost obligatory cryptic loss of Ikaros, required for the normal B cell maturation. Although similar losses were found in lymphoid blast crisis the genetic background of the transformation in CML is still poorly defined. We used Significance Analysis of Microarrays (SAM) to analyze comparative genomic hybridization (aCGH) data from 30 CML (10 each of chronic phase, myeloid and lymphoid blast stage), 10 Ph(+)ALL adult patients and 10 disease free controls and were able to: (a) discriminate between the genomes of lymphoid and myeloid blast cells and (b) identify differences in the genome profile of de novo Ph(+)ALL and lymphoid blast transformation of CML (BC/L). Furthermore we were able to distinguish a sub group of Ph(+) ALL characterized by gains in chromosome 9 and recurrent losses at several other genome sites offering genetic evidence for the clinical heterogeneity. The significance of these results is that they not only offer clues regarding the pathogenesis of Ph(+) disorders and highlight the potential clinical implications of a set of probes but also demonstrates what SAM can offer for the analysis of genome data.
Keywords
Introduction
Array CGH (aCGH) has shown itself to be a mature technology capable of detecting genomic gains and losses at a resolution of at least 250 base pairs. Clearly at this resolution there will be masses of data that will challenge the analyst, particularly in the light of the general variance of the genome from individual to individual—so called copy number variations (CNV) and knowledge that the function of a substantial part of the genome is still unknown hence referred to as ‘orphan’ or ‘predicted'. It is also clear that genomic copy number aberrations (CNA) associated with diseased cells are likely to interfere with transcriptional pathways and affect gene function.
Significance Analysis of Microarrays (SAM) was developed by Tusher 1 as a straightforward way of comparing data sets using an internally generated false discovery rate (FDR) as the criterion for the identification of probes that significantly differ between 2 or more classes. It has been successfully used to analyse gene expression data and is now routinely applied.2,3
Philadelphia positive malignant disorders are a clinically divergent group of leukaemias with a unique identifying feature, the BCR/ABL1 fusion gene, usually resulting from the chromosome rearrangement t(9;22)(q34;q11) or its variants, that leads to constitutive expression of an aberrant tyrosine kinase. These include chronic myeloid leukaemia (CML) and de novo acute leukaemia of both myeloid Ph(+)AML and lymphoid origin Ph(+)ALL. The latter two disorders are clinically aggressive and therapy challenging even in the era of the powerful tyrosine kinase inhibitors. CML is a multistage progressive disorder which if untreated inevitably ends as fatal acute myeloid or lymphoid blast transformation. The latter, from which it has been reported to differ karyotypically, is usually clinically indistinguishable from Ph(+)ALL the most common type of ALL in adults. 4 Although non-random chromosome changes may accompany disease progression in CML, the genetic background of the malignant transformation from the benign chronic phase (CP) to acute leukaemia (blast crisis, BC) is poorly understood. Recent whole genome screening identified a spectrum of cryptic aberrations associated with disease progression5,6 sometimes even present at the onset of CML. 7 Also similar investigations of Ph(+)ALL in both children and adults identified recurrent cryptic loss of Ikaros, required for the normal B cell maturation 8 in addition to the known deletions of the p16 (CDKN2A) gene.
These findings led us to look in CML and Ph(+) ALL for imbalances in DNA sequences significantly associated with the disease stage and lineage origin. We used array CGH data obtained from 40 anonymous bone marrow samples comprising 10 CML chronic phase, 10 CML lymphoid blast phase, 10 CML myeloid blast phase, 10 Ph(+)ALL from the UKALLXII(R) trial [9] and 10 peripheral blood samples from disease free individuals. Of the ALL samples 5 had t(9;22)(q34;q11) as a sole cytogenetic abnormality, one was Ph negative but BCR/ABL positive, 3 showed hyperdiploid karyotype (HEH) but none showed aberrations detectable by G banding in the short arm regions of chromosome 7 and 9 (Table 1). The presence of the BCR/ABL1 fusion gene was confirmed in all samples by qPCR and/or FISH (D-FISH probe, Vysis, USA) as reported previously. 9 All Ph(+)ALL and 5 out 10 CML blast crisis samples had established B cell immunophenotype.6,10
Summary of chromosome and fish results for the Ph(+)ALL cases.
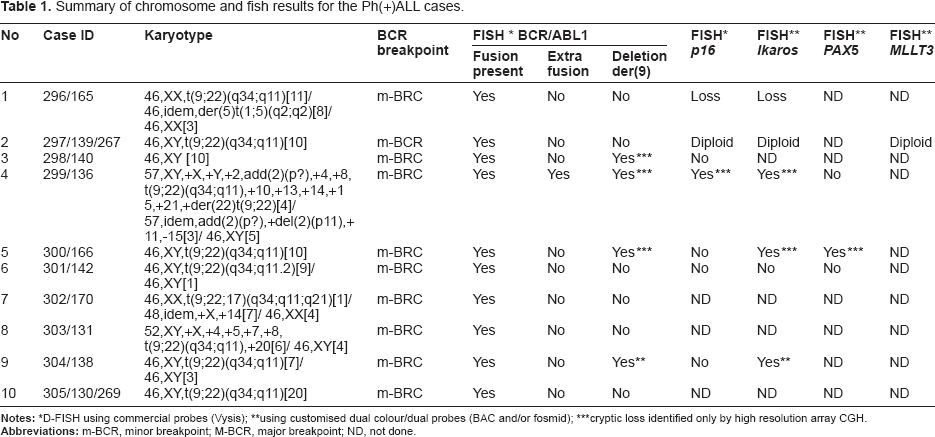
D-FISH using commercial probes (Vysis);
using customised dual colour/dual probes (BAC and/or fosmid);
cryptic loss identified only by high resolution array CGH.
Having identified genome regions of potential interest, ranked in order of significance, out of the thousands of array results, it is then a challenge to design further experiments to evaluate their contribution to the biology of the BCR/ABL positive disease.
Materials and Methods
Array CGH analysis was performed as described previously. 6 Briefly, Agilent (Wilmington, DE, USA) oligonucleotide arrays were hybridized following the manufacturer's protocol. 500 ng genomic test DNA was extracted from either peripheral blood or BM samples. Sex mismatched pooled DNA from peripheral blood mononuclear fraction of 6–8 disease free individuals (Promega, UK) was used as reference. Customized Agilent oligonucleotide arrays comprising 8 × 15 k probe sets per slide were designed from an analysis of active loci (hot spots) in the CML BC genome corresponding to pairs of probes that exceeded a 3SD threshold in 3 or more CML BC samples from a previous study. 5 Probes were selected to cover regions at ~1 k intervals except where the presence of repetitive sequences disallowed the inclusion of reliable probes.
The arrays were scanned, features extracted and the data analyzed using an Agilent scanner and Mathematica software (http://www.wolfram.com). In addition, all samples had been subjected to whole genome screening using 105 K Agilent oligonucleotide arrays as part of published study. 6
The emergence of high throughput technology such as microarrays raises a fundamental statistical issue relating to testing hundreds of hypotheses thus rendering the standard P value meaningless. 11 The False Discovery Rate (FDR) concept is an alternative to the P value. Tusher 1 described such a method: Significance Analysis of Microarrays (SAM) and the implementation due to Chu et al has been incorporated by J Craig Venter Institute into their suite of ‘MeV’ routines. 12 SAM uses permutations of sample labels to estimate the FDR. We report the application of SAM for 5,000 permutations setting the median number of false significant probes to zero, for the supervised analysis of the myeloid and lymphoid blast crisis, chronic CML, Ph(+)ALL and control samples. Firstly, after removing data for the sex chromosomes, we constructed a table defining the log fluorescence ratio (FR) for each locus and assigned classes eg, Lymphoid blast phase CML (L) or Myeloid blast phase CML (M); Ph(+)ALL (ALL); Chronic phase CML (C); Control (Ctrl); Male (m) or Female (f). We chose a two class unpaired test and applied SAM to ask if there were any probes that were uniquely associated with either classification. All genome addresses are derived from build 35 (March 2006) of the Human Genome.
Results
Genomic difference between lymphoid and myeloid lineages
We applied SAM to seek correlations between genome imbalances and clinical presentation. We asked which probes were significantly involved in the discrimination between lymphoid and myeloid lineages using the classes of myeloid and lymphoid CML BC as a model. Altogether we identified 489 significant probes, the top 100 of which were restricted to the TCR, IKZF1 and IgH genomic regions. Figure 1 shows cluster analysis of the 40 most significant probes indicating losses occurring at genome address between 105,405,310 and 105,518,122 mbp in the IgH region, between 38,287,976 and 38,315,044 mbp in TCR and between 50,385,101 and 50,429,250 mbp in the sequences of IKZF1 (Table 2). Lymphoid samples including Ph(+)ALL clustered together displaying losses (Fig. 1 on the left), while the myeloid blast crisis and chronic CML samples formed a separate cluster with the control samples (Fig. 1 on the right). We noted that 3 samples sat at the myeloid/lymphoid borderline and that some of the control samples showed losses in the TCR region.
The 40 most significant probes differentiating between lymphoid and myeloid lineages.
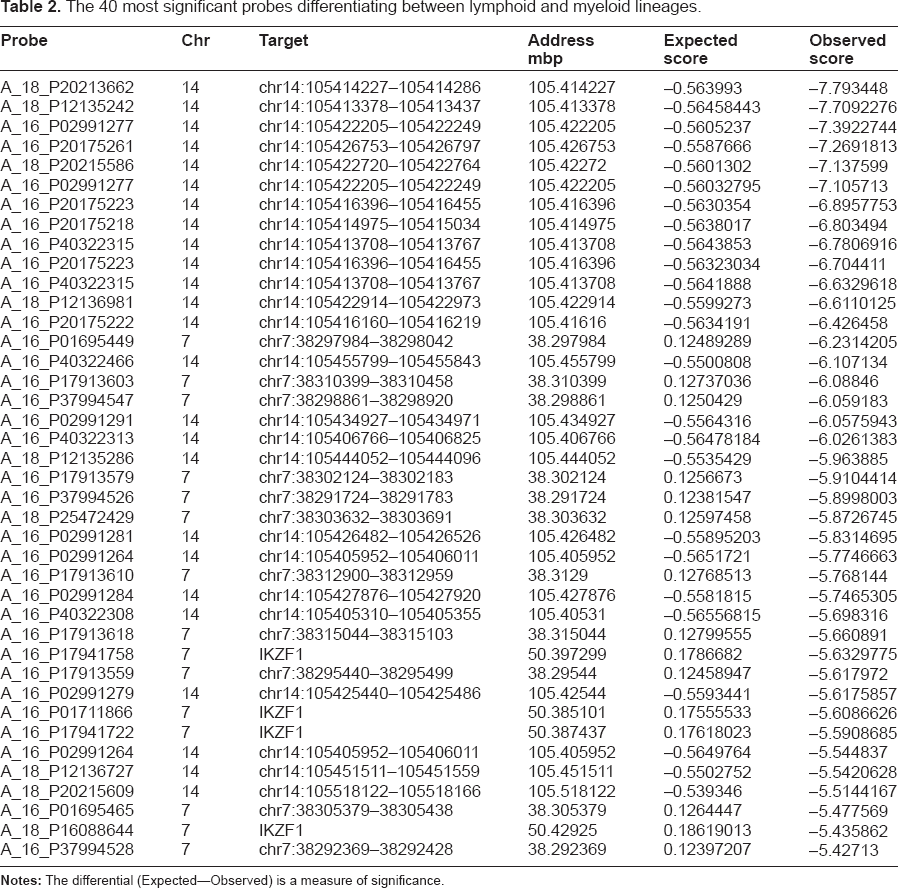

Top 40 most significant probes from a cluster analysis of 489, distinguishing lymphoid and myeloid BCR/ABL1 positive genomes.
Comparison of CML lymphoid blast crisis and Ph(+)ALL
84% of the 155 probes differentiating lymphoid blast crisis CML and Ph(+)ALL map to one of two regions of the short arm of chromosome 9, namely 9p21.3–p21.2 and 9p24.1–p23, the latter housing genes PTPRD and MLLT3 among others. A hierarchical cluster analysis shows that five of the 10 Ph(+)ALL cases form a cluster of gains (Fig. 2, in red) although cytogenetic revealed no structural or numerical changes of 9p (Table 1). In contrast, half of the 10 CML BCL cases formed a cluster with extensive genome loss (in green) that had been previously shown to be complex by G-banding and 105 K oligonucleotide array. 6 See Figure 2 and Table 3.
The 40 most significant probes differentiating between Ph(+)ALL and lymphoid blast crisis.
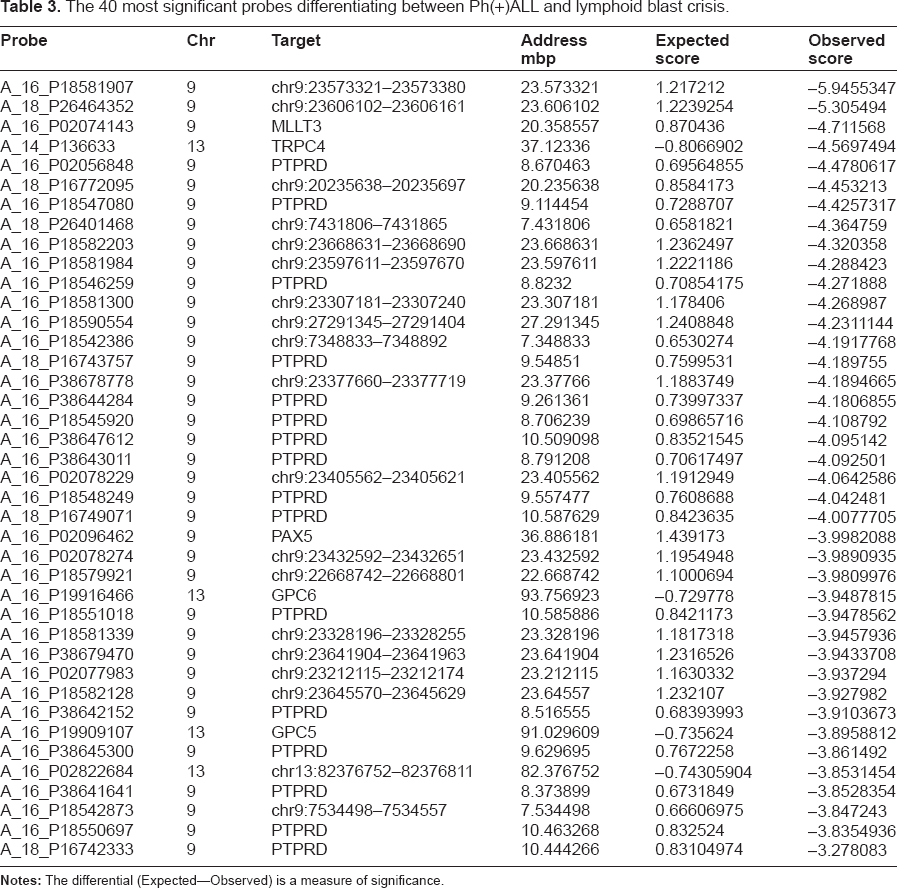

Identification of probes discriminating between ph positive acute lymphoblastic leukemia and CML lymphoid blast transfomation.
Since many of the significant probes fell on chromosome 9p we repeated the analysis excluding all chromosome 9 loci. The top 10 of 80 probes meeting our significance threshold are revealed by cluster analysis (see Fig. 3 and Table 4). Associated with these loci are known genes such as PDEA4 (cAMP-diestarase) in band 19p13.2 and GSTT1 in band 22q11.23, one of the most commonly reported polymorphic marker (CNV) in man. Genome loss (in green, Fig. 3) dominates the profile of 6 out 10 Ph(+)ALL samples. Surprisingly 5 of the latter cases (297, 299, 300, 301, 303) exhibit gains in the chromosome 9p21–p24 region (Fig. 3, heat map A).
The 40 most significant probes differentiating between Ph(+)ALL and lymphoid blast crisis excluding chromosome 9 probes.


Ph positive all with gains at 9p21–p24 share common losses elsewhere in the genome.
It is suggested from the heat maps in Figures 2 and 3 that the Ph(+)ALL samples split into two groups, 5/10 cases showing dominant amplification of loci in the chromosome 9p region and losses elsewhere in the genome, while the remainder (5/10) lack recurrent genome imbalances. However, we were unable to detect any consistent differences in the two groups of Ph(+)ALL samples from an inspection of their chromosome status (see Table 1).
In summary, SAM analysis revealed that while the lymphoid blast stage CML and Ph(+)ALL samples share common losses within the IGH, TCR, and Ikaros gene regions together with loci within the 9p21–p24 region, they form separate clusters at other sites on the genome thus suggesting that these acute malignant conditions may represent separate biological entities.
Discussion
Whilst huge progress has been made in the analysis of the genome and the identification of genes associated with malignant disease, there is still much work to be done evaluating the function of coding and non-coding regions 13 . We have identified numerous short 60 mer sequences that appear to play a significant role in the evolution of Philadelphia positive hematological malignancy. We offer no explanation of their function, but provide convincing evidence that their involvement is not a random event.
SAM is used for the analysis of expression arrays to classify samples into groups according to phenotype using false discovery rate (FDR) as a test for significance.1,14 Here we use SAM to study DNA from a cohort of CML and Ph(+)ALL patients to identify sequences that may help to distinguish between these Philadelphia positive diseases and enlighten their pathogenesis.
Numerous software packages are available for the detection of genomic gains and losses across a range of array technologies, reviewed by Shah,
15
but high-resolution array data presents special problems as typified by a wide variance making detection of small features complicated. Individual signals are rarely if ever considered to be significant on their own but only in the context of a contiguous collections of gains and losses. However if an individual locus is compared across a number of similarly processed arrays, the probability of a random single signal exceeding a 3SD threshold for
We designed a high resolution array (~1 kb intervals) designed to explore regions of the genome shown previously at low (33 kb) resolution to display gains or losses in a cohort of 35 samples from CML patients in blast phase 6 . This necessitated sacrificing large areas of the genome to concentrate on these areas for detailed inspection. Using this set of ~15,000 genetic loci enabled us to confirm that lymphoid phenotypes formed a single group characterized primarily by unique deletions within the IgH regions consistent with an early VDJ rearrangement as part of the B cell receptor formation occurring in per-B cells together with loss of the TCR gamma sequences also indicating gene rearrangement. Loss of whole or part of the IKZF1 (Ikaros) gene is the third most common feature in the genome profile of these cases. In contrast with a typical CNV that could affect any part of the IgH gene on 14q32.33 the deletions identified by us always involve the sequences 105.41–105.48 mbp and are almost universally accompanied by deletions in the TCR region of chromosome 7. Both IgH and TCR sequences are usually excluded from aCGH analysis as they are reported to be CNVs. We have demonstrated that these deletions are consistent throughout the sample set, suggesting that they are disease specific. These findings could be explained by a chain of events initiated by BCR/ABL1 that leads to compromised V(D)J recombinase machinery thus creating clonal populations of early B-cell progenitors with cross lineage rearrangements. 6 Mullighan et al in their poster presentation “Genome wide analysis of Genetic Aberrations in Chronic Myeloid Leukemia” (Mullighan et al, http://ash.confex.com/ash/2008/webprogram/Paper5715.html) reported results from SNP analysis of 90 CML samples of which 9 were diagnosed as lymphoid blast crisis. This study could not find any genomic features that could differentiate between BC/L and Ph(+)ALL. In contrast we were able to reveal genomic differences in these clinically similar conditions.
Many of the ‘significant’ probes that distinguish between Ph(+)ALL and BC/L cluster within chromosome 9p21 region, which harbours the CDKN2A/B gene, the loss of which has long been associated with both haematological and solid tumours and shown to result from RAG impairment. 8 Since 5 out of the 10 BC/L CML cases were found to carry imbalances of the short arm of chromosome 9, it is possible that some probes in this location were lost ‘by association’ and not involved in discriminating between these two diseases. However, we found other loci that did discriminate between Ph(+)ALL and BC/L CML. For example, while half of the BC/L cases had deletions in chromosome 9p, half of the of Ph(+)ALL showed gains at these loci as shown in Figure 2. Omitting chromosome 9 records and reanalyzing the data, the same five Ph(+)ALL samples showed significant losses in 80 loci from other chromosomal locations (Fig. 3). Taken together the tandem CNA—gains at 9p with recurrent losses elsewhere in the genome offer a way to differentiate a Ph(+)ALL from CML lymphoid BC. Whilst we recognize that single aberrant 60 mer sequences could easily be dismissed as random events, the fact that there are more than 80 widely distributed probes not associated with morphological or cytogenetic anomalies but associated with a significant minority of the Ph(+)ALL samples is worth consideration. Further work is required to explore the possible role of these genome aberrations. In conclusion, SAM results offer clues regarding the pathogenesis of BCR/ABL1 positive disorders and furthermore identifies a sets of probes with diagnostic potential.
Authors’ Contribution
CG carried out the SAM analysis and co-wrote the manuscript; EPN designed the study and co-wrote the manuscript.
Funding
This work has been supported by LRF grant No. 05098, Jean Coubrough Charitable Trust Project to Dr. EP. Nacheva.
Competing Interests
EPN's institution has received grants from the Leukemia Research Fund and the Jean Coubrough Charitable Trust, and travel or study support from Cytocell. Research funding was also received from operation of a clinic associated with UCL. CG discloses no conflict of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgements
We are grateful to Dr. Adele Fielding, Prof. Letizia Foroni and Prof. Anthony Moorman for valuable comments on the manuscript and providing samples; to Dr. Diana Brazma who carried out the aCGH and performed FISH tests and Mrs. J. Howard-Reeves for karyotyping and FISH analysis.