
Abstract
Motivated by the frustration of translation of research advances in the molecular and cellular biology of cancer into treatment, this study calls for cross-disciplinary efforts and proposes a methodology of incorporating drug pharmacology information into drug therapeutic response modeling using a computational systems biology approach. The objectives are two fold. The first one is to involve effective mathematical modeling in the drug development stage to incorporate preclinical and clinical data in order to decrease costs of drug development and increase pipeline productivity, since it is extremely expensive and difficult to get the optimal compromise of dosage and schedule through empirical testing. The second objective is to provide valuable suggestions to adjust individual drug dosing regimens to improve therapeutic effects considering most anticancer agents have wide inter-individual pharmacokinetic variability and a narrow therapeutic index. A dynamic hybrid systems model is proposed to study drug antitumor effect from the perspective of tumor growth dynamics, specifically the dosing and schedule of the periodic drug intake, and a drug's pharmacokinetics and pharmacodynamics information are linked together in the proposed model using a state-space approach. It is proved analytically that there exists an optimal drug dosage and interval administration point, and demonstrated through simulation study.
Introduction
The past three decades have seen spectacular advances in our understanding of the molecular and cellular biology of cancer. However, data suggest that the overall success rate for oncology products in clinical development is ~10%, and the cost of bringing a new drug to market is over US $1 billion. 1 Oncology drug development is such an expensive and prolonged process, typically, a new drug requiring on average 10 years.2,3 New tools are needed to accelerate the drug discovery process and increase productivity.4,5 While producing information both at the basic and clinical level is no longer the issue, 6 the effective integration of data and knowledge from many disparate sources will be crucial to future cancer research.7,8 Systems biology approaches promise to have a profound impact on medical practice by bringing together efforts from cross disciplinary scientists and permitting a comprehensive evaluation of underlying predisposition to disease, disease diagnosis, disease progression and disease treatment.9–11
While providing the right drug for the right patient is very important, finding the right dose for each patient is also critical but tricky. 12 Finding a dose and dose range of a drug candidate that are both efficacious and safe is a fundamental objective through the drug discovery process. 13 Dose finding happens throughout the long process of drug discovery, from non clinical development to multi-phase clinical trials. Even after the drug is approved and available on the market, new drug doses are still studied carefully and the level of investigation depends on responses observed from the general patient population. When necessary, dose adjustment based on post-marketing information is still a common practice. However, it is extremely expensive and difficult to get the optimal compromise of dosage and schedule through empirical testing. Modeling and simulation analysis, which can evolve and be continuously updated throughout different stages to incorporate relevant new data, will help to make crucial decisions earlier, with more certainty, and at lower cost, and hence can add value in all stages of drug development.5,14
The complexity of cancer itself and the heterogeneity of therapeutic responses may make dosing study more complicated. For example, most anticancer agents have wide inter-individual pharmacokinetic (PK) variability and a narrow therapeutic index. 15 Recent works have shown that many patients who are currently being treated with 5-fluorouracil (5-FU) are not being given the appropriate doses to achieve optimal plasma concentration. Of note, only 20%–30% of patients are treated in the appropriate dose range, approximately 40%–60% of patients are being underdosed, and 10%–20% of patients are overdosed. 16 Traditionally, the standard approach for calculating 5-FU drug dosage, as with many anticancer agents, has been done by normalizing dose to body surface area (BSA), which is calculated from the height and weight of the patient; 16 however, studies have shown that this is inadequate. 17 For example, dosing based on BSA is associated with considerable variability in plasma 5-FU levels by as much as 100-fold,15,17 and such variability is a major contributor to toxicity and treatment failure. 16 Since there are many factors collaboratively affecting drug effect variability, 18 a general approach is needed to facilitate quantitative thinking to drug administration regimens. Drug dosing regimens could be tailored to each individual patient based on feedback information from the treatment. One challenge of such modeling is how to link relevant biomarkers 19 or surrogate endpoints to treatment outcome as feedback information in order to give valuable dosing suggestions.
Traditional design of the dosing regimen based on achieving some desired target goal such as relatively constant serum concentration may be far from optimal owing to the underlying dynamic biological networks. For example, Shah and co-workers 20 demonstrate that the BCR-ABL inhibitor dasatinib, which has greater potency and a short half-life, can achieve deep clinical remission in CML patients by achieving transient potent BCR-ABL inhibition, while traditional approved tyrosine kinase inhibitors usually have prolonged half lives that result in continuous target inhibition. A similar study of whether short pulses of higher dose or persistent dosing with lower doses have the most favorable outcomes has been carried out by Amin and co-workers 21 in the setup of inactivation of HER2-HER3 signaling. For best results, models should be selected based on the underlying mechanism of drug action. For example, detailed dynamic signal transduction models are needed to accommodate target-receptor interaction and feedback loops for analyzing dosing effect in the above examples.
Computational systems biology is emerging as a valuable tool in therapeutics to address these challenges.10,22–25 This approach provides functional understanding of disease-drug interaction and marks a shift from the traditional “black-box” approach. In this study, a general methodology incorporating dynamic drug pharmacology information into drug therapeutic response modeling using computational systems biology is proposed. The process begins with building a quantitative model of a biological system. Then, by incorporating related pharmacology information relevant to the target system, a new computational model under drug perturbation can be built. We believe that with the help from the theoretical modeling proposed in this study and through an iterative process with experimentalists to refine the model, the proposed methodology has the potential to supply better recommendations for dosage and frequency.
Modeling
A good model should be based on a sound understanding of the biological problem, hold a realistic mathematical representation of the biological phenomena, and possess a tractable solution. 26 A biological interpretation of the deductions resulting from such a model can yield non-intuitive insights, as well as provide a predictive framework, 10 a vital issue in cancer treatment. In recent years it has become clear that carcinogenesis is a complex process, both at the molecular and cellular levels.25,27 Modeling biological systems to develop computer models of disease that can be used to understand disease mechanisms and to test in silico approaches for treating disease is a key issue in moving forward. Recent mathematical advances have made it more feasible to model cancer from a mathematical viewpoint. There are numerous works modeling cancer at multiple levels and scales, ranging from molecules to cells to tissues.7,28 For example, multi-scale models have been developed that can capture interactions across different spatial and temporal scales. 8 A number of researchers have recommended hybrid or hierarchical systems to combine the strengths of both discrete and continuous approaches.8,29,30
Biological systems are naturally nonlinear; however, purely nonlinear continuous models of biological systems can be too large and complex for simulation and analysis. On the other hand, a linear continuous model or a fully discrete approximation of the model can sometimes lose crucial and pertinent information. 31 Hybrid systems 32 provide a rigorous foundation for modeling biological systems at desired levels of abstraction, approximation, and simplifications. 33 For example, systems that exhibit multi-scale dynamics can be simplified by replacing certain slowly changing variables by their piecewise constant approximations. Additionally, sigmoidal nonlinearities are commonly observed in biology and the corresponding models often use sigmoidal functions. These can be approximated by discrete transitions between piecewise-linear regions. In some instances, nondeterministic upper and lower bounds are more useful than deterministic approximations because they capture all critical behavior of the system.33–35 The hybrid systems model encapsulates a broad space of models and systems. For example, the Lac operon system has been well studied both experimentally and using continuous models.36,37 A hybrid model and use of a reachability algorithm were validated by comparison with experimental data and continuous models. 38 Other biological hybrid systems analyzed in similar ways include the Delta-Notch decision process,39,40 genetic regulatory networks of carbon starvation, 41 nutritional stress response 42 in E. coli, and our previous work on drug effect modeling under genetic regulatory networks. 43 In this study, we adopt hybrid systems models to accommodate the hybrid nature of disease progression and therapeutic responses. Specifically, a tumor growth model under drug perturbation is studied to demonstrate how to integrate diverse data and ultimately predict outcomes for clinical purposes.
Tumor growth model using hybrid systems
Cancer research has been a fertile ground for mathematical modeling. 44 A number of mathematical tumor growth models have been reported in the literature, reflecting different paradigms. Empirical models use mathematical equations to describe the tumor growth curve without in-depth mechanistic description of the underlying physiological processes. Initially, models were used to conceptualize the simple exponential growth of solid tumors.45–47 Subsequently, sigmoidal functions such as logistic, Verhulst, Gompertz, and von Bertalanffy were used for the description of reduced growth in the later stages as the tumor cells outgrew their blood supply, producing central necrosis.48–51 A drawback of this model class is that it is not straightforward to predict modification of the growth curve under drug perturbation. Functional models, conversely, are based on mechanistic descriptions of biological processes underlying tumor growth. Such models require a set of assumptions involving cell cycle kinetics (proliferating vs. quiescent cells) and biochemical processes, such as those related to antiangiogenic and/or immunological responses.52,53 Owing to the biological complexity they try to capture, these models have a much larger number of parameters compared with the empirical models. Hence, in addition to the standard tumor growth measurements, further data are needed, such as flow cytometry analysis and measurements of biochemical and immunological markers, to avoid identification problems due to the over parametrization. 53 The situation becomes even more complex when the effect of treatment with an anticancer drug is considered on account of the incomplete knowledge of the mode of action in vivo.
It is important to realize that all models have limitations, including those in oncology: simple models may produce insights and describe existing data, but they risk oversimplification and oversight of critical variables; on the other hand, it is generally difficult to fit functional models versus experimental data since over parametrization can be avoided only if further “microscopic” observations are available. Hence, it is a challenge to achieve a correct balance between empirical and functional models. In this paper, we adopt a model that is a compromise between empirical and mechanism-based approaches.54,55 The model is based on a system of ordinary deferential equations that link the dosing regimen of a compound to the tumor growth in xenograft mice, with tumor growth in untreated animals being described by exponential growth followed by a linear growth phase. In treated animals, the tumor growth rate decreases proportionally to both drug concentration and the number of proliferating tumor cells. It relies on a few identifiable parameters, the estimation of which requires only the data typically available in the preclinical setting. There are two parameters related to drug effect: c(t), the drug concentration, and k2 a constant measuring drug potency. 55 In their later study, 56 good correlation was achieved with a novel approach proposed to predict the expected active dose in humans from the studies mentioned above.54,55
Although modeling in tumor growth has attracted a lot of attention, most of the aforementioned efforts have not explored the impact of drug effects. There is some work assuming the system is at steady state, which means that the concentrations of active drugs at the active site are constant. In some drug effect models,55,57,58 drug effect is assumed to be related to drug concentration and number of tumor cells; however, if we would like to compare drug effects for different dosing regimens to consider issues as whether we give patient frequent small or infrequent large drug dosage given fixed total drug intake, more realistic and dynamic drug effect models are needed. This study proposes a model dynamically linking disease progression, in which hybrid systems are adopted to accommodate disease progression and therapeutic responses. Specifically, we adapt the tumor growth model proposed in 54 to hybrid systems model to accommodate the tumor growth dynamics in different stages and augment it with a drug effect model related to PK and pharmacodynamics (PD). In this proposed framework, PK and PD are linked by a state-space approach, where drug concentration will fluctuate between dosages and drug efficacy will change with drug concentration. Our main aim is to model drug effect on tumor growth for different treatment dosing regimens given related pharmacology information.
Unperturbed growth model (without drug treatment)
Following the same biology setup as Magni et al, 54 unperturbed and perturbed growth models are formulated to model tumor growth dynamics without treatment and with treatment, respectively. Tumor growth is modeled by an exponential growth phase followed by a linear growth phase for the unperturbed growth model. A hybrid systems model is proposed to accommodate tumor growth dynamics in different stages. It takes the form

where w u denotes unperturbed tumor weight, β1 and β0 are parameters characterizing the rates of exponential and linear growth. s+(·) is the unit step function defined by

s−-(·) = 1 – s+(·), and θ w is the corresponding threshold value at which tumor growth switches from exponential to linear growth. To assure the continuity of derivatives in Equation (1) at θ w , θ w = β0/β1 can be derived. Given current progress in tumor growth modeling, the tumor growth characteristics might be quite different in different situations. The proposed model based on hybrid systems can be extended to accommodate more complicated cases, such as more growth stages with different growth rates.
Perturbed growth model (with drug treatment)
All the tumor cells are assumed to be proliferating in the unperturbed model. With drug treatment, it is assumed that cells affected by drug action stop proliferating and pass through different stages characterized by progressive degrees of damage and eventually they die. 55 A transit compartment model is used for the cells’ progression to death under drug treatment:




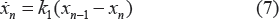


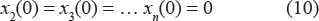
where x1 indicates the portion of proliferating cells within the total tumor weight w
p
with drug treatment. x1(t) will go through exponential and then linear growth similar to the unperturbed tumor model, where β1 and β0 denote the respective growth parameters. In these equations, w
p
is the total tumor weight, represented by the sum of cells in the various stages, and w0 is the tumor weight at the inoculation time (t = 0). Since not all cells are proliferating, the linear growth rate is slowed down by the ratio of the proliferating cells over the total tumor cells x1/w
p
. The model assumes that the drug elicits its effect related to the number, x1, of proliferating cells and
Drug treatment model
The basis of clinical pharmacology is the fact that the intensities of many pharmacological effects are functions of the amount of drug in the body and, more specifically, the concentration of drug at the effect site. 59 For a long time, PK and PD had been considered as separate disciplines; however, the information provided by these disciplines is limited if regarded in isolation. On one hand, PK is characterized as what the body does to the drug, and it denotes the concentration-time course of drugs in different body fluids. On the other hand, PD is assessed as what the drug does to the body, and it characterizes the intensity of effects resulting from certain drug concentrations at the assumed effect site. In order to describe the time course of drug effect in response to different dosing regimens, the integrated PK/PD model is indispensable, which builds the bridge between these two classical disciplines of pharmacology. 60 Following each dosing regimen, instead of a two dimensional dose-concentration (PK) and concentration-effect (PD) relationship, our proposed approach enables a description of a three dimensional dose-concentration-effect relationship. Specifically, PK and PD are linked through a state-space approach to facilitate the description and prediction of the time course of drug effects resulting from different drug administration regimens.
PK/PD modeling is an active research area in pharmacology5,59 Application of such concepts has been identified as potentially beneficial in all phases of preclinical and clinical drug development.61,62 This work is our first attempt in the direction of quantitative drug effect modeling. Although we make some assumptions about concentration-effect and dose-concentration curves in this paper, the methodology proposed is flexible enough such that many specific PK/PD data can be accommodated in the proposed framework. If the mathematical formulation becomes too complicated and theoretical analysis is not possible, an extensive simulation study can be carried out for available PK/PD data.
Periodic drug intake: pharmacokinetics (PK) model
We consider a periodic drug intake scenario. One could use a detailed theoretical or empirical pharmacokinetic description of time dependent drug concentration at the site of action in a simulation study. We prefer to keep the model mathematically tractable so that we can perform a strict theoretical analysis and thereby gain insights. Thus, we assume the concentration has exponential decay. Since we are using hybrid systems, the PK model can be extended to include more complicated cases, such as the case where the drug concentration will first exponentially increase, then slowly change (equilibrium), and then exponentially decrease.
63
The model used for drug intake and concentration levels is illustrated in Figure 1. We denote the period of drug intake for the two cases as τ1 and τ2, respectively. Without loss of generality, it is assumed that τ1 = Mτ2, where M > 1 is an integer. It is also assumed that u1(kτ1) = ζ1 = Mu2(lτ2) = Mζ2, where k and l are non-negative integers, and ζ1 and ζ2 are dosages in cases 1 and 2, respectively. This means that, in the long run, the patient takes the same total drug amount in both cases. It is assumed that the concentration level of the drug at the effect site follows exponential decay during each period, ie,

The concentration level of drug under periodic drug intake. Two cases are shown: (1) large dose with longer period; (2) small dose with shorter period.
Drug efficacy and potency: pharmacodynamics (PD) model
The PK model provides the concentration time course resulting from the administered dose and the continuous description of concentration will serve as input function for the PD model, which relates the concentration to the observed effect. Generally, the magnitude of pharmacological effect increases monotonically with increased dose, eventually reaching a plateau level where further increases in dose have little additional effect. 13 The classic and most commonly used concentration-effect model is the Hill equation, 64 also called the sigmoidal Emax model 65 or logistic model. 66 The relationship between the concentration of the drug candidate and its effect is most often nonlinear. In some cases, the curve even looks like a “roller coaster”, which is referred to as the “double Hill Model”. 67 One common method is to replace certain slowly changing variables by their piecewise linear approximation. In this study, we use hybrid systems to approximate the sigmoidal Emax PD model (see Fig. 3). The Emax model has the general form:
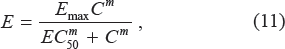
where Emax is the maximum effect, C is the concentration, EC50 is the concentration necessary to produce 50% of Emax, and m represents a sigmoidity factor or steepness of the curve.
We assume a threshold of concentration below which the drug candidate is ineffective (such dose is often called the minimum effective dose (MinED)). We assume another threshold called the maximum effective dose (MaxED), above which there is no clinically significant increase in pharmacological effect. We use a linear curve to approximate the concentration-effect curve between MinED and MaxED. We assume that the drug effect coefficient
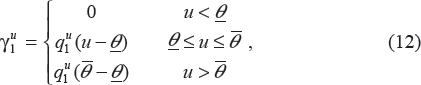
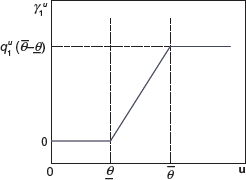
The Concentration-effect curve.
where

Sigmoidal Emax model (m = 4), and approximation by our PD model.
Drug effect analysis
Based on the proposed perturbed growth model (Eqs. (3) to (7)), the drug effect is related to the number of proliferating tumor cells x1 and drug effect coefficient

where

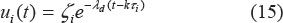
where ui(t) is the drug concentration level at the assumed effect site.
State-space analysis
In our proposed model, there are both continuous quantitative changes (eg, the drug concentration level) and discrete transitions (eg, PD model). As is common in hybrid systems, there are both continuous and discrete states. The entire state space may be divided into different domains according to the value of the discrete state. When the quantitative change of the continuous state meets certain criteria, it will cause a discrete transition from one domain to another. Specifically, after each drug intake, the drug concentration at the effect site is dynamically changing following the PK model, with the changing concentration falling into different ranges (domains). The tumor growth dynamics will change according to different PD model at each domain. A state space and trajectory plot of the state of the proliferating tumor growth and drug concentration level under periodic drug intake are illustrated in Figure 4. There are five domains in the state space, with D1 D3, D5 not being transient.

The trajectory of the state (tumor weight growth and drug concentration level) under periodic drug intake
The figure shows the case when the drug is effective and the initial drug concentration level is larger than the upper threshold
Depending on the initial drug intake, the state trajectory may start from different domains. For example, if the initial conditions are x1 = x1(kτ
i
) and u
i
= ζ
i
>
Case 1: state trajectory starts from domain D5
We define t1 as the traveling time from the initial condition to the boundary between D5 and D3, and t2 as the traveling time from the initial condition to the boundary between D3 and D1. The traveling time within D3 is therefore t2 – t1. Since we are considering the case that state trajectory starts from domain D5, the initial conditions are x1 = x1(kτ
i
) and
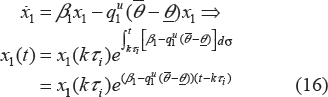

In order to reduce x1, we need
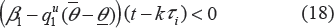
this implies

–D3 (from time t1 to t2, ie, t1 ≤ t ≤ t2):


In order to reduce x1 we need

–D1 (from time t2 to (k + 1)τ i , ie, t2 ≤ t ≤ (k + 1)τ i ):
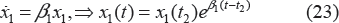

Since the drug dosage is below the effective level (
For the drug to be effective, both the inequalities (22) and (19) must be satisfied; however, they are just loose bounds. We could deduce the necessary and sufficient condition for the effectiveness of the drug by expressing the inequality x1((k + 1)τ) ≤ x1(kτ) in terms of the dose period τ and unit dose ζ, so that after each treatment period the number of proliferating tumor cells x1 will decrease. When the initial conditions are x1 = x1(kτ
i
) and


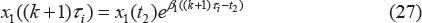


and can be simplified to


For the drug to be effective, we need Ψ < 0, so that the number of proliferating tumor cells x1 will decrease following each period of drug intake.
Case 2: state trajectory starts from domain D3
When the drug dosage is below
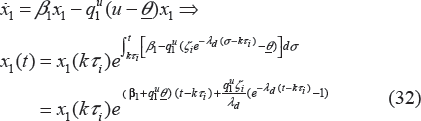

–D1 (from time t2 to (k + 1)τ i ):
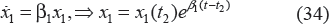
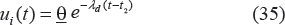
The equations governing the state trajectory from time kτ i to time (k + 1) τ i are given by

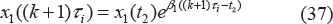

This can be simplified to


For the drug to be effective, we need Ψ < 0, so that the number of proliferating tumor cells will decrease following each period of drug intake.
Tumor growth minimization
We have proved mathematically that the reduction of the number of proliferating tumor cells, defined as x1((k + 1)τ i ) – x1 (kτ i ), is a strictly convex function 68 of time interval τ i . The detailed proof is given in the Appendix A. This implies that the function of tumor size reduction, which should be negative for a successful treatment, has a “U” shape and has a unique global minimum point, 68 where x1((k + 1)τ i )–x1 (kτ i ) < 0 is the smallest that corresponds to the maximum reduction in tumor size (where |x1((k + 1)τ i )–x1(kτ i )| is the largest).
Results
In order to validate the analytical results on drug effect for different dosing regimens, we firstly perform numerical simulations using predefined parameters to validate the analytical results. Then we proceed with parameter estimation on synthetic data sets generated based on the experimental study.54,55 This second step is critical to facilitate the use of hybrid mathematical model to biologist. We also demonstrate that similar conclusion on drug efficacy region can be drawn based on the synthetic data sets generated using the parameters from real experiments.
Simulations using predefined parameters
In order to validate the analytical results on drug effect, we firstly perform numerical simulations using MATLAB/SIMULINK, based on the detailed transit compartment model presented from Eqs. (3) to (7). The cells affected by drug action stop proliferating and pass through four different stages, x1, x2, x3, and x4, characterized by progressive degrees of damage, where x1 indicates the portion of proliferating cells and w
p
is the total tumor weight. Specifically, the parameters in the simulation are β1 = 1.0, β0 = 0.2, k1 = 1.0, θ
w
= 40. For the PD model, we follow Eq. (12) and set the parameters as
Observation 1: To compare the effect of different dosages and frequencies given a certain total drug intake, we define the density of drug intake as

τ = 5, ζ = 15, Ψ = 0.7.
τ = 8, ζ = 24, Ψ = –0.24.

τ = 15, ζ = 45, Ψ = 1.5.
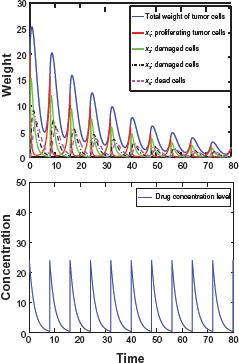
Figures 5–7 show the responses of tumor under three dosing regimens (all with same total drug intake α = 3.0). The left figure (Fig. 5) corresponds to the small frequent dosing, the right figure (Fig. 7) corresponds to the large infrequent dosing, and the case of intermediate dosing in between (Fig. 6). Other parameter setting for the above figures: β1 = 1.0, β0 = 0.2, k1 = 1.0,
We observe that the changes of total tumor weight w p follow similar patterns with the changes of the number of proliferating cells x1, which confirms our theoretical analysis. Moreover, the results clearly show that dosing regimens play a critical role in disease treatment, even when the total drug in take remains the same. Both the small frequent dosing (Fig. 5) and large in frequent dosing case (Fig. 7) do not reduce the tumor size effectively. Only in the case with moderate dosage and interval (Fig. 6), are both the number of proliferating cells and the total tumor weight reduced effectively. At the same time, the results demonstrate what is predicted in the analytical results: we need Ψ < 0 so that the tumor will degrade following each period of drug intake.
Observation 2: To further verify that the change of tumor weight between treatments, Δx1 = x1((k + 1) τ) – x1(kτ), is a strictly convex function of time interval, we plot the percentage of tumor weight change versus dosing period τ in Figure 8 for different total drug intakes with α = 2.8, 2.9, 3.0, 3.1, 3.2. For each curve with fixed total drug intake, it can be seen that Δx1 is indeed convex, and an optimal choice of drug administration can be made based on the point of maximum tumor reduction. There exist some “sweet spots” (defined as “drug efficacy regions”) of drug administration that will satisfy the condition Ψ < 0. The three special dosing cases (Figs. 5–7, with α = 3.0) can be tested in the curve and it is easily confirmed that only the moderate dosage and interval case with τ = 8 falls into the drug efficacy region. Furthermore, Figure 8 illustrates the tradeoff between efficacy and toxicity. When the total drug intake α increases, the drug efficacy region gets larger accordingly. At another extreme, the drug efficacy region may not exist when α gets too small.
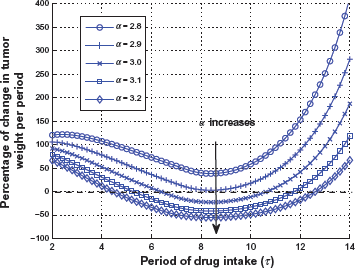
The percentage of tumor weight change change vs. τand total drug intake α, where decay rate λ d is same).
Observation 3: There are many factors that affect drug response, inter-individual PK variability being one of them. Thus, it is important to check how different PK parameters change drug effect. In this study, we plot the percentage of tumor weight change versus τ for different PK decay rates (λ d = 0.42, 0.44, 0.46, 0.48, 0.5) in Figure 9. It is observed that the effect of PK (specifically, the decay rate λ d ) on tumor reduction is significant. When the drug decay is slow, say λ d = 0.42, the tumor weight will decrease much faster than when drug decay is fast, say λ d = 0.5. This confirms our hypothesis that the drug effect is closely related to the PK parameters, which is one reason for the heterogeneity of therapeutic responses. Hence, if we could estimate inter-individual PK variability based on accurate measurement and interpretation of drug concentration in biological fluids and perform corresponding therapy assessment to model disease progression, then it is possible that we could adjust dosing regimens during treatment based on such feedback information for each individual following the methodology presented in this study, thereby improving the drug's therapeutic effect.

The percentage of tumor weight change vs. τand decay rate λ d , where total drug intake is the same (α = 3).
Simulations using synthetic data generated from experimental study
In this part of the study, synthetic data sets are produced from experimental study conducted by Magni, Simeoni and et al54,55 firstly. Then we perform parameter estimation based on the synthetic data sets using nonlinear least square method. 69 Finally we show that similar observations on drug efficacy can be obtained using these synthetic data sets.
Generation of synthetic data
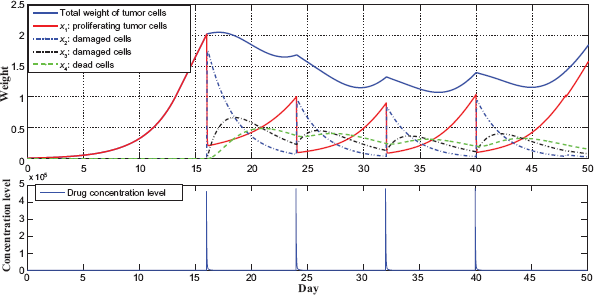
Although the experimental data sets54,55 are not publicly available, the authors54,55 provided the parameter values such that the tumor growth model in their papers matches their experimental data very well. Hence, we perform numerical simulation using the model given in the study54,55 to produce synthetic data sets. Specifically, the PK data (drug plasma concentration) is generated by using the model of c(t) given by Equations (17)–(19) on page 138 of Magni et al 54 and the corresponding parameter values given in Table 2 on page 140 of Magni et al. 54 The tumor growth data during the entire treatment process is generated by firstly using the unperturbed model given by Magni et al 54 for the first 15 days, then using the perturbed model given by Magni et al 54 with the input from the PK data for 32 days (day 16 to day 47). Then the treatment is stopped from day 48 and on. In order to model the drug effect due to different drug plasma concentration, we also include the sigmoidal Emax model as given by Eq. (11) in our numerical simulation. The SIMULINK block diagrams for generating PK data and the entire treatment process are given in Figures 10 and 11, respectively. The generated synthetic data of a typical run is plotted in Figure 12 for the case of taking drug every day from day 16 to day 47.

SIMULINK block diagrams for generating PK data.
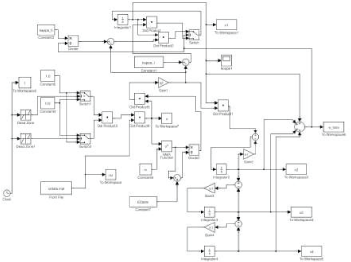
SIMULINK block diagram for generating tumor growth data during the entire treatment process.

The tumor growth data from a typical run for the case of taking drug every day from day 16 to day 47.
It is observed that the tumor grows exponentially for the first 15 days, then the weight of proliferating cells x1 dropsfrom 2 to 1.25 when drug is taken from day 16 to day 47. At the same time, the entire tumor starts growing slower and eventually reduced and stabilized. During each day, due to the PK/PD profile, x1 reduces sharply when the initial drug concentration is high, then x1 starts to increase because the drug concentration decreases exponentially.
Parameter Estimation
Because of the nonlinear nature of the model, we applied nonlinear least square method 69 for parameter estimation. Specifically, we use the “nlinfit“ function in MATLAB statistical toolbox. nlinfit returns the least square parameter estimates, ie, it finds the parameters that minimize the sum of the squared differences between the observed responses and their fitted values. It uses the Gauss-Newton algorithm with Levenberg-Marquardt modifications for global convergence. The detailed steps for parameter estimation is illustrated below.
Use nlinfit function to estimate the exponential growth parameter β1 based on the measurements of the tumor size from the first 15 days and the unperturbed tumor growth model.
Now by plugging in the estimated values of β1,
Note that since x
i
(t) cannot be experimentally measured, it is not feasible to estimate the time-varying parameter
The plot of nonlinear least square curve fitting for parameters β1 and k1 are given in Figures 13 and 14, respectively. It can be seen that β1 can be accurately estimated without much error. The true value is β1 = 0.349 and the mean of the estimates is
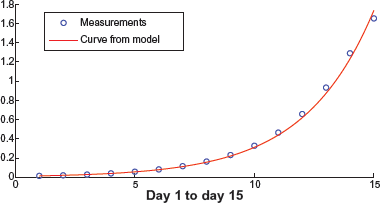
Curve fitting for parameter β1.

Curve fitting for parameter k1.
Drug efficacy under different administrations
In order to obtain insights on the drug efficacy under various dosage and frequency schedules, we study the drug effect for 5 different dosing regimens with the same total drug intake, specifically, (1) once per day with the dosage given by Magni, Simeoni and et al54,55; (2) double dosage given every two days; (3) 4 times dosage given every 4 days; (4) 8 times dosage given every 8 days; (5) 16 times dosage given every 16 days; respectively, during the 32-day (day 16 today 47) treatment process. The detailed plots for case (1) are given in Figure 12, the detailed plots for the rest cases are given in Figure 16 to 19 from Appendix B. The results are compared in Figure 15. It is observed that taking drug every 4 to 8 days seems to reduce the tumor the most, and without much oscillations. Of course, which dosing regimen to choose is depending on many other practical considerations, including toxicity. It is demonstrated that although the total drug intake during the 32-day treatment process remains the same, different dosage and frequency schedules do have significant impact on the tumor growth, which is consistent with what we obtained analytically and observed before using predefined parameters.
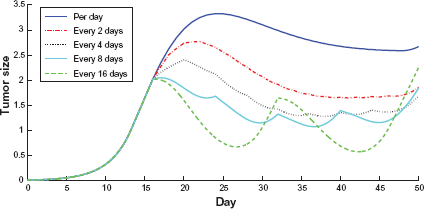
Comparison of tumor growth under different dosage and frequency schedule from day 16 today 47.

The tumor growth data from a typical run for the case of taking double dosage every two days from day 16 today 47.

The tumor growth data from a typical run for the case of taking 4 times dosage every 4 days from day 16 today 47.
The tumor growth data from a typical run for the case of taking 8 times dosage every 8 days from day 16 today 47.

The tumor growth data from a typical run for the case of taking 16 times dosage every 16 days from day 16 today 47.
Observation 4: Through this study based on synthetic data generated from experimental study,54,55 it is clear that the parameters can be estimated by the measurements of tumor weights along the treatment process. This would enable the proposed hybrid system model to be applied to study drug effects in real-world experiments. We believe that it is feasible to refine the model with the experimentalist through an iterative process, then such model can be used to predict the drug effect and provide better recommendation for different dosing regimens.
Conclusion
A proof-of-concept study of quantitative drug effect modeling has been carried out using hybrid systems. Specifically, the PK/PD data are linked together with tumor growth dynamics in our analysis of therapeutic effects. This is a small step towards quantitative modeling of drug effect and we have kept the examples simple so that they are mathematically tractable and valuable insights can be obtained from the analytical results. For example, we have demonstrated that drug effect is closely associated with different dosing regimens and individual PK/PD characteristics, and the simulation results match the theoretical analysis. Although the examples in this paper are simple, the proposed framework for quantitative modeling of drug effect is flexible enough to be able to incorporate many practical PK/PD data as well as different models for tumor growth if desired. Of course, when more complicated PK/PD data and tumor growth models are used in the proposed framework, analytical results may not be attainable and one may have to rely on a simulation tool built on the proposed framework to obtain the drug effect for different dosing regimens and individual PK/PD characteristics.
Disclosures
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributor ship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.
Footnotes
Acknowledgements
Xiangfang Li has been supported by the National Cancer Institute (2 R25CA090301-06). The authors would like to thanks anonymous reviewers’ valuable suggestions to make this a better paper.
Proofs of Theorems
Theorem 1. Changes of the number of proliferating cells x1 dominate the changes of all the tumor cells under drug treatment.
Proof. Firstly we express the equations of the tumor growth model under drug treatment (Eq. (3) to (7)) in matrix form
Let
It can be seen that at any given time, the solution of the matrix Eq. (41) is completely determined by the eigenvalues of matrix A. To be more specific, suppose that w p < θ w , then A is reduced to
It can be deduced that the diagonal terms,
Theorem 2. The change of proliferating tumor size in one treatment, defined as x1((k + 1)τi) — x1(kτi), is a strictly convex function of time interval ti.
Proof. We define the reduction of proliferating tumor size in one treatment as
To show that Δx1 is a strictly convex function of time interval τ
i
, we need to prove that the second derivative of Δx1 with respect to τ
i
is positive for any τ
i
. We show the case where the trajectory starts from domain D5; similar techniques can be used to show it for the other case where the trajectory starts from domain D3. When the drug dosage is high enough, ie,
Now the first derivative of Δx1 with respect to τ i is given by
where
Since both x1(kτ i ) and eΨ are positive, we only need to show that
which is equivalent to
It is straightforward to verify that the above inequality (49) is always true since a1 > 0, a3 > 0, β1 > 0. Then the theorem follows.
Detailed plots for Section “Parameter Estimation”
In order to demonstrate that drug effect is depending on the dosing regimens given the same total drug intake, detailed drug effect plot is showed in this appendix. While Figure 12 provides the case for taking drug every day from day 16 to day 47, the following figures are the detailed plots for: double dosage given every two days (Fig. 16); 4 times dosage given every 4 days (Fig. 17); 8 times dosage given every 8 days (Fig. 18); 16 times dosage given every 16 days (Fig. 19); respectively, during the 32-day (day 16 to day 47) treatment process.