
Abstract
Living on earth, we are exposed to ultraviolet (UV) light as part of the solar radiation. UVB spectrum light exposure contributes to the development of skin cancer by interacting with pyrimidine pairs to create lesions called cyclobutane pyrimidine dimers. If these lesions are not removed by nucleotide excision repair, they often give rise to C to T transition mutations. Based on these observations, a bioinformatics approach was used to predict the vulnerability of human protein coding genes to UVB induced loss of function mutations. This data was used to evaluate in depth those genes associated with malignant melanoma. In addition, we demonstrate a method of genetically engineering genes that significantly improves resistance to UVB loss of function mutations.
Introduction
Skin cancers are the most common primary type of cancer. The three most common primary types of cutaneous cancer are squamous cell carcinoma, basal cell carcinoma, and malignant melanoma. Melanoma is of particular interest since it tends to occur in younger adults, and has a relatively poor prognosis if metastasis has occurred. 1 Human epidemiological studies as well as lab animal experiments indicate that UV exposure is an important etiological factor in all three types of skin cancer. 2
The spectrum of solar UV light can be divided by wavelength into UVA, UVB, and UVC. UVB spectrum light exposure is thought to explain 80%–90% of the mutagenic effects of sunlight. 2 DNA sequence analysis has revealed the target and effect of UVB exposure, which has come to be known as the ‘UVB signature’ mutation. 3 The targets of UVB in DNA are pyrimidine pairs containing cytosine, ie, CC, CT, or TC. Following UVB exposure, these pyrimdine pairs give rise to either C->T point mutations or CC->TT dinucleotide changes. 2
The mechanism by which UVB exposure causes this pattern of mutagenesis is partially understood. UVB energy can be absorbed by pyrimidines and can result in two new intrastrand covalent bonds formed between adjacent pyrimdines. 4 This structure is known as a cyclobutane pyrimidine dimer (CPD). How the CPD gives rise to the observed mutations is unclear. One model supposes that a DNA polymerase introduces the incorrect nucleotide when using the CPD containing strand as the template. Another model supposes that at least one of the cytosines in the CPD are methylated and that the CPD structure increases the basal spontaneous deamination of 5-methylcytosine to thymidine. 2
However, not every CPD that forms will necessarily give rise to a mutation. CPD's create a distortion in the DNA that can potentially be recognized by the nucleotide excision repair (NER) enzymes. 4 These enzymes can excise part of the strand flanking a CPD and use the complementary strand as a template to replace the excised sequence. The importance of this repair pathway is highlighted by Xeroderma pigmentosum patients who exhibit increased frequency of skin cancers. 2
To sum up, when cells are exposed to UVB, UVB signature mutations may occur. 3 If these genetic alterations perturb key regulators of the cell cycle or apoptosis, skin cancer may develop. Several genetic loci have been associated with malignant melanoma including Tumor Protein 53 (TP53), Cyclin-dependent Kinase 4 (CDK4), Cyclin-dependent Kinase Inhibitor 2A (CDKN2A), Melanocortin 1 Receptor (MC1R), Microphthalmia-associated Transcription Factor (MITF), v-Kit Hardy-Zuckerman 4 Feline Sarcoma Viral Oncogene Homolog (KIT), v-Raf Murine Sarcoma Viral Oncogene Homolog B (BRAF), Neuroblastoma RAS viral Oncogene Homolog (NRAS), B-cell Lymphoma 2 (BCL-2), Apoptotic Protease Activating Factor 1 (APAF1), v-AKT Murine Thymoma Viral Oncogene Homolog (AKT), Phosphatase and Tensin Homolog (PTEN), and FK506 Binding Protein 12-Rapamycin Associated Protein 1 (FRAP1). 1
CDK4 is a protein kinase that phosphorylates Retinoblastoma (Rb) to enable G1-S cell cycle progression.
7
Familial melanoma has been associated with lesions in CDK4.
8
The
MC1R is a seven transmembrane G protein coupled cell surface receptor for melanocyte-stimulating hormone (MSH-alpha).
10
Receptor activation results in a signaling cascade resulting in expression of MITF and other genes neccesary for eumelanin synthesis.
11
MC1R polymorphisms are associated with melanoma.
12
MITF is a transcription factor that is a master regulator of melanocyte development, survival and melanogenesis.
13
Amplification of
The tyrosine kinase KIT is required for the developmental migration and proliferation of melanocytes.
15
Upon binding its ligand Stem-cell Factor, KIT activates a mitogen-activated protein kinase (MAPK) signaling cascade.
16
Activating lesions in
NRAS is a member of the RAS family of plasma membrane bound small G proteins that can be activated by receptor tyrosine kinases. 18 When in its GTP bound form, RAS can activate RAF. 19 RAF is a protein kinase that is part of a MAPK signaling cascade that promotes cellular proliferation. 20 NRAS is mutated in 33% of primary melanomas, and associated with increased activity. 18 BRAF is mutated in about two thirds of melanomas, typically causing increased activity. 1
BCL-2 is primarily found in the outer membrane of mitochondria where it acts to prevent release of apoptosis mediators from the mitochondrial intermembrane space.
21
The expression of BCL-2 may correlate with melanoma progression and
APAF1 forms apoptosomes with cytosolic cytochrome c.
24
This complex activates caspase-9, resulting in programmed cell death.
FRAP1, also known as mammalian Target of Rapamycin (mTOR), is a protein kinase that is found in two different complexes called mTORC1 and mTORC2. 29 mTORC1 promotes protein translation, favoring cell proliferation. 29 mTORC2 activates AKT by phosphorylation. 29 Increased mTOR activity was observed in 73% of melanomas. 30
Despite the known importance of UVB sites in the etiology of skin cancers, there has been little work characterizing their distribution in the human genome. One goal of this work was to have a genome wide analysis of UVB sites with respect to coding sequences. Furthermore, we investigated the degree to which these UVB sites would be deleterious if mutated. In addition, we explored the relationship between UVB sites and genes implicated in malignant melanoma. One supposition is that these coding sequences would have an overabundance of UVB sites that may predispose them to mutation. Another possibility is that the UVB sites found in these coding sequences are present in a sequence context that is more deleterious than that of the average gene.
Finally, we propose a novel approach for genetically modifying skin cancer susceptibility. Given that UVB sites are the target of UVB mutagenesis, decreasing the frequency of these sites should decrease the susceptibility of a coding sequence to UVB mutation. We propose a method for decreasing the frequency of UVB sites while maintaining the wildtype primary amino acid sequence by the rational use of synonymous codon substitution. In this work, we bioinformatically predict the extent to which this method can improve UVB resistance of coding sequences.
Methods
An algorithm was developed which first identifies potential UVB sites in human protein coding sequences, individually introduces UVB signature mutations at these sites, and then predicts the consequence to the protein product. The UVB signature target site is defined as any of the dinucleotides CC, CT, and TC (and GG, AG, and GA considering the change may occur on the complementary strand). The algorithm identifies every UVB site in translated exonic genomic sequences. In addition, UVB sites may potentially be found at the junctions of exons, therefore, when extracting an exon from genomic sequence, we also took one additional nucleotide from each side of an exon in the analysis to not miss those instances where the UVB site straddled the junction. These flanking nucleotides were trimmed after screening for UVB sites in the coding sequence.
The algorithm then individually creates a series of UVB single point mutants of the coding sequence by introducing single transition mutations (C->T or G->A) at each site. For CC and GG dinucleotide sites, there would be two separate single transition mutant sequences generated. In addition, UVB doublet mutants of coding sequences were created at dinucleotides CC (and GG considering the change may occur on the complementary strand) by introducing two transitions mutations (CC->TT or GG->AA). Doublet mutants at CC and GG dinucleotides were not created if they straddled two codons (corresponding to phase 3 position of the first codon and phase one position of the subsequent codon) because this information was already captured in the two single mutants created.
For each mutant, the BLOSUM62 amino acid substitution matrix was then used to quantitatively assess the consequence of the altered codon when translated. 31 BLOSUM62 uses a log odds ratio score and awards negative scores to unfavorable amino acid substitutions. 32 The sum score for all unfavorable mutations for a gene was computed to get an overall amino acid change score representing the UV susceptibility of the protein coding gene (Fig. 1). A perl script was written to carry out this algorithm.

Schematic representation of algorithm to assess UVB sensitivity. At left, boxes are drawn around UVB sites. Below the double stranded DNA is the single letter amino acid sequence for each codon of the upper strand. In the middle, nucleotides that have undergone mutation are shown in bold. The translated sequence is shown below the DNA. The BLOSUM62 substitution matrix score for each new amino acid compared to its original amino acid is shown at the bottom. At right, the sum of the negative BLOSUM62 scores is given as the total amino acid change score for this sequence.
Version 37 of the human genome chromosomal genbank flat files were obtained for analysis (http://www.ncbi.nlm.nih.gov/Ftp/). A total of 32,357 coding sequences (CDS) were analyzed. 95 coding sequenes were not analyzed because they were incomplete fragments. Due to our inclusion of flanking nucleotides in screening for UVB sites, the calculation of length of a coding sequence for the purpose of estimating UVB frequency, was defined as the sum of all coding sequence nucleotides including the stop codon + 2 · (number of exons that contained coding sequence).
Genes were optimized for UVB resistance by selective synonymous codon substitution. The wildtype primary amino acid sequence was maintained but used the following codons substituted for the original codons: F = > TTT, L = > TTA, I = > ATT, M = > ATG, V = > GTA, S = > TCA, P = > CCA, T = > ACA, A = > GCA, Y = > TAT, STOP = > TAA, H = > CAT, Q = > CAA, N = > AAT, K = > AAA, D = > GAT, E = > GAA, C = > TGT, W = > TGG, R = > CGT, G = > GGT.
GC percentage, length, and dinucleotide composition were calculated for each coding sequence. The output from the perl script was parsed into an Excel spreadsheet for ease of viewing. The data was imported into R for statistical analysis using the Bioconductor package multtest.33,34
Chi squared tests were used to estimate
The expected number of deleterious single mutation sites in a CDS was calculated by multiplying the number of UVB sites in each CDS by 0.298. The number 0.298 is the mean frequency of a single mutation being deleterious among all coding sequences.
The expected number of deleterious double mutation sites in a CDS was calculated by multiplying the number of CC and GG UVB sites by 0.679. The number 0.679 is the mean frequency of a double mutation being deleterious among all coding sequences.
To analyze average severity of single deleterious mutations,-a-control-sample-of-the-first-65000 negative BLOSUM62 single amino acid scores were collected from chromosome 5, which had a mean of –2.14. This set of scores was used for comparison of the single mutation negative scores from each melanoma related coding sequence two tailed Student's t-test with equal variances.
Results
We investigated the frequency of UVB sites in human coding sequences. At the outset, we expected UVB sites to be found 3/8 (or 38.75%) of the time for any dinucleotide of sequence considered. This assumption was based on probability with equal frequency (25%) of the four nucleotides. This expectation was modified based on the average GC composition of the coding sequences of 53.48% to arrive at a 39.18% frequency of a UVB site at any given dinucleotide to be expected by chance. Interestingly, the observed mean frequency of UVB dinucleotides among all coding sequences was 44.5% ± 5.2%, which was significantly greater than expected (
On average, only about 30% of the potential single transitions would result in an unfavorable amino acid substitution (Table 1). Considering doublet UVB mutations at positions 1–2 or 2–3 of codons, about 68% of these would give rise to unfavorable amino acid changes. Unfavorable mutations at doublet sites also tended to be more deleterious substitutions with a mean BLOSUM62 score of –2.69 per residue change compared to –2.16 at single unfavorable sites.
UVB characteristics of human coding sequences.

The number of UVB sites in human coding sequences could be important for susceptibility to UVB mutagenesis. We counted the number of UVB sites in human coding sequences and observed that the number increases linearly as a function of the length of the coding sequence (R 2 = 0.988) (Fig. 2). In addition, based on our model, the calculated amino acid change score grows more negative in a linear fashion with respect to length (R 2 = –0.939) (data not shown). Thus, longer open reading frames are predicted to be more susceptible to UVB. However, neither the number of UVB sites nor the amino acid change score correlated with the GC percentage of the coding sequence (R 2 = –0.005 and R 2 = –0.063) (data not shown).

Scatterplot of the number of UVB sites as a function of length of the coding sequence.
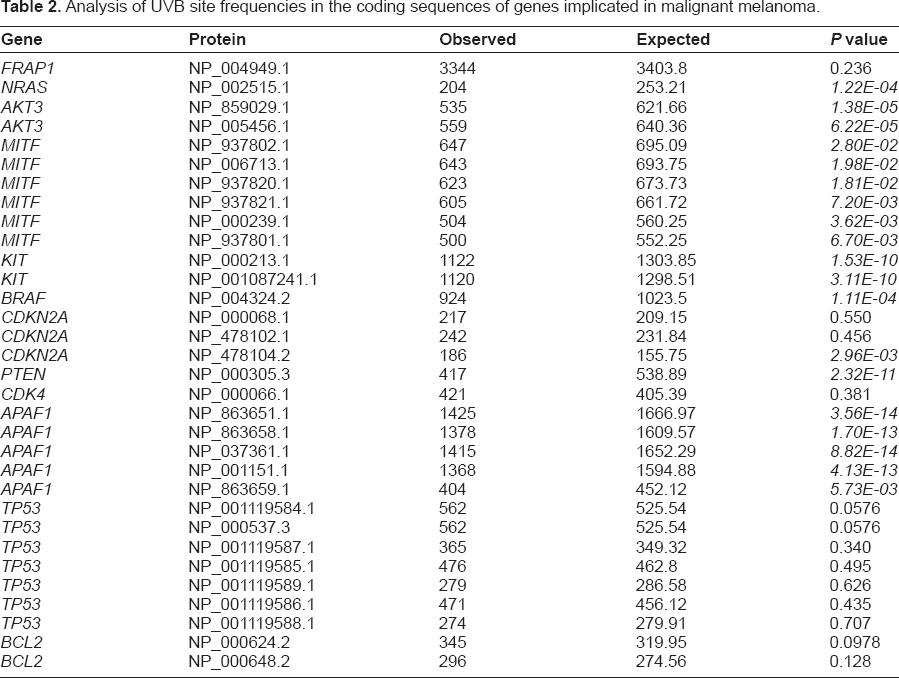
We investigated whether genes associated with malignant melanoma would have a signficantly different number of UVB sites than would be expected from the average frequency of UVB sites. Seven of the genes had significantly fewer than expected UVB sites, while four were not significantly different from the average gene. One isoform of CDK2NA was found to have a significantly increased number of UVB sites (Table 2).
Analysis of UVB site frequencies in the coding sequences of genes implicated in malignant melanoma.
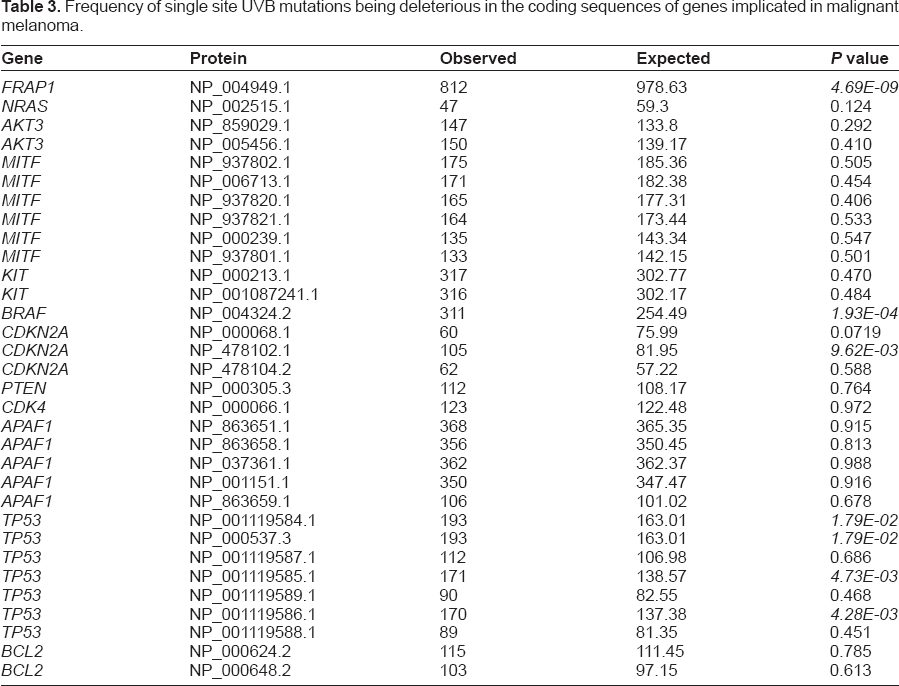
It could be that a greater proportion of the UVB sites in genes implicated in malignant melanoma could give rise to deleterious substitutions if mutated. The predicted proportion of UVB site mutations causing a deleterious change was evaluated in the genes linked to malignant melanoma (Tables 3 and 4). A subset of those coding sequences did have a significantly-increased proportion of sites predicted to produce unfavorable changes, including four of seven coding sequences from
Frequency of single site UVB mutations being deleterious in the coding sequences of genes implicated in malignant melanoma.
Frequency of deleterious double site UVB mutations in the coding sequences of genes implicated in malignant melanoma.

We studied whether the UVB sites in coding sequences of genes implicated in melanoma would on average cause more deleterious changes than those found in the average coding sequence. This would be reflected by more negative BLO-SUM62 scores. The predicted mean severity of UVB deleterious mutations was compared in genes linked to melanoma and a large control group (Table 5). Mutations at UVB sites in the coding sequences of
Mean severity of deleterious single mutations in the coding sequences of genes implicated in malignant melanoma.

Finally, we explored the use of rational synonymous codon substitution to alter the coding sequence without changing the primary amino acid sequence in order to decrease the predicted vulnerability to UVB (Table 6). This method was found to decrease the number of UVB sites by almost 50% and improve the amino acid change score for the coding sequences of all genes associated with malignant melanoma by about 25%.
Synonymous codon substitution can decrease predicted UVB vulnerability of genes implicated in malignant melanoma.

Discussion
Despite the importance of UVB sites in skin cancer formation, the extent of these targets and their potential for deleterious mutations has not been previously investigated on a genome-wide basis. We found that these sites are quite prevalent in human coding sequences and are predicted to have signficant deleterious potential. However, it was suprising that the majority of the time, the predicted consequence of any one single transition mutation at a UVB site is not unfavorable. It is interesting to note that the average coding sequence has more UVB sites than would be expected at random. This may be a consequence of codon usage biases or it may reflect an adaptive mechanism to facilitate UVB molecular mutation.
As UVB is a major causative factor in skin cancer and leads to UVB signature mutations, we hypothesized that the sequence properties of genes may influence their likelihood to suffer from UVB induced loss of function mutations. That is, certain genes would have sequences that either contain more UVB targets or the targets tend to occur in a more sensitive context that would lead to unfavorable mutations in either greater frequency or severity or some combination thereof. Surprisingly, most genes associated with melanoma have a fewer than expected number of UVB-sites. This may reflect a selective process for these genes to avoid UVB mutagenesis. However, our data suggests that all three mechanisms may be at play depending on the coding sequence. Some coding sequences have an increased number of UVB sites, some have an increased frequency of UVB sites that are deleterious and some tend to have more deleterious changes on average when mutated. These qualities may be useful in screening for additional genes important in melanoma, to identify additional diagnostic and therapeutic molecular targets.
Metastatic melanoma is associated with a 10 year survival rate of less than 10%. 36 In addition, the frequency of melanoma is increasing and over 60,000 new cases occur each year in the United States. 37 Therefore, the need for new methods for primary prevention of melanoma or in its treatment are greater than ever. We have described a new method for decreasing the susceptibility of coding sequences to UV, much like sunscreen for genes. Although currently preclinical, cutaneous gene therapy has promise. 38 As gene therapy advances, genetically engineering decreased vulnerability of a gene to UVB may be useful therapeutically in the treatment of this devastating disease.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Supplementary Data
Collection of output files from perl script are available at http://bengal.missouri.edu/kale0a.
Perl script: Mel15.pl.
Excel file of data in a tabular format: mel_table.xls.
Footnotes
Acknowledgments
The authors would like to thank the anonymous reviewers for their comments and suggestions.