
Abstract
Informatics and computational design methods were used to create new molecules that could potentially bind antiapoptotic proteins, thus promoting death of cancer cells. Apoptosis is a cellular process that leads to the death of damaged cells. Its malfunction can cause cancer and poor response to conventional chemotherapy. After being activated by cellular stress signals, proapoptotic proteins bind antiapoptotic proteins, thus allowing apoptosis to go forward. An excess of antiapoptotic proteins can prevent apoptosis. Designed molecules that mimic the roles of proapoptotic proteins can promote the death of cancer cells. The goal of our study was to create new putative mimetics that could simultaneously bind several antiapoptotic proteins. Five new small molecules were designed that formed stable complexes with BCL-2, BCL-XL, and MCL-1 antiapoptotic proteins. These results are novel because, to our knowledge, there are not many, if any, small molecules known to bind all three proteins. Drug-likeness studies performed on the designed molecules, as well as previous experimental and preclinical studies on similar agents, strongly suggest that the designed molecules may indeed be promising drug candidates. All five molecules showed “drug-like” properties and had overall drug-likeness scores between 81% and 96%. A single drug based on these mimetics should cost less and cause fewer side effects than a combination of drugs each aimed at a single protein. Computer-based molecular design promises to accelerate drug research by predicting potential effectiveness of designed molecules prior to laborious experiments and costly preclinical trials.
Introduction
Conventional drug design techniques are based on trial-and-error testing using cells or animals. High-throughput screening for chemicals with desired bioactivities requires specialized labs that make the process costly. With a growing number of known experimental structures of target molecules, computational methods have been used successfully to supplement and speed up drug discovery. Computer-based molecular design combines methods of informatics, medicine, and biophysics. This cross-disciplinary field has accelerated drug research by predicting the potential therapeutic effectiveness of designed molecules prior to laborious experiments and costly preclinical trials. In addition, computational modeling has led to discoveries of structures of novel small molecules. In this work, informatics and computational design were used to create and evaluate new small molecule mimetics that could potentially promote death of cancer cells.
Apoptosis is an important cellular process that causes death of damaged cells. 1 Its malfunction can lead to cancer development and poor response to conventional chemotherapy. 2 Cellular proteins from the BCL-2 family are crucial for apoptosis..3,4 Understanding their interactions is vital for anticancer drug design. 5 Proteins from the BCL-2 family can be either prodeath (proapoptotic) or prosurvival (antiapoptotic). Antiapoptotic proteins, such as BCL-2, BCL-XL, and MCL-1, share homology in three to four conserved BH peptide domains (BH1, BH2, BH3, and BH4)..6,7 Proapoptotic proteins, such as BAX, BAK, BIM, BAD, and BID, share homology only in the BH3 domain.
Cellular damage stimulates prodeath stress signals. After being activated by the stress signals, proapoptotic proteins can bind antiapoptotic proteins, thus allowing apoptosis to proceed. Stress signals are sensed by the BH3-only proteins called “activators”, which can activate proapoptotic BAX and BAK proteins..8,9 This leads to the mitochondrial membrane deterioration.10,11 and commits the cell to apoptosis. The, so called, “sensitizer” proteins allow apoptosis to proceed by preventing the binding of antiapoptotic proteins to activators. 12 Propagation of death signals is hindered by antiapoptotic proteins. 13 Antiapoptotic protein BCL-2 resists apoptosis mainly by binding activators, thus precluding their activation of BAX and BAK..14,15 Cancer cells can avoid death signals by overexpressing antiapoptotic proteins, inhibiting proapoptotic activators, or precluding BAX and BAK activation. 16 These can also lead to poor chemotherapy response. 17 Disabling of antiapoptotic proteins is often needed for apoptosis to proceed. 16 BH3 mimetics are designed molecules that mimic the functions of BH3-only proapoptotic cell proteins. 18 The BH3 mimetics can help destroy cancer cells by inhibiting antiapoptotic proteins and specifically targeting pathways that allow survival of cancer cells. 19
Previously designed small molecule ABT-737 [Protein Data Bank 20 (PDB) entry: 2YXJ] allows BAX and BAK activation by binding BCL-2, BCL-XL, and BCL-W and disrupting their complexes with proapoptotic proteins. 21 ABT-737 was originally designed by combining two molecules of medium affinities to obtain one molecule of high affinity. 4
Previous studies revealed that ABT-737 was deadly to many cancer cells. 4 It was reported that it makes cells with overexpressed BCL-2 ready for apoptosis at a much higher degree than those with overexpressed BCL-XL, although their affinities for ABT-737 are similar. 4 The reason for this is unclear, which suggests that interactions of ABT-737 with these proteins are still not well understood. ABT-737 is an effective single agent in cancers with low or absent MCL-1. 4 However, ABT-737 is not efficient in cancer cells with overexpressed MCL-1. Preclinical studies show that ABT-737 could easily induce apoptosis in many cancer cells if MCL-1 is eliminated or suppressed. 22 Using ABT-737 in combination with an agent that can target MCL-1 holds great clinical promise.
Obatoclax is a designed molecule (ChemDB database 23 compound ID (CID):16681698) that can supplement the activity of ABT-737. Although Obatoclax has a lower affinity than ABT-737 for BCL-2, BCL-XL, and BCL-W, 24 it can successfully prevent MCL-1/BAK binding. 25 Another small molecule, Antimycin A, is able to suppress cancer growth if used at high concentrations. 26 A combination of drugs targeting several antiapoptotic proteins may be necessary to overcome resistance to apoptosis in some cancers..27,28 A designed molecule, ABT-702 (ChemDB database CID: 3965212), shows anti-inflammatory actions in cells. 29 To the best of our knowledge, ABT-702 has not been studied for anticancer applications. Its complexes with BCL-2, BCL-XL, and MCL-1 are not known. However, using ABT-702 as a starting template, we were able to design new potential mimetics that bonded BCL-2, BCL-XL, and MCL-1 proteins and showed drug-likeness.
Computational studies of small molecule mimetics can be done using several commercial and open-access docking programs and servers, such as ArgusLab, 30 GOLD, 31 AUTODOCK, 32 and ZDOCK. 33 Commercial GOLD docking program has been previously used to successfully study interactions between BCL-2 and several small molecules. 34 It was found that the binding energies of stable complexes formed by BCL-2 and C33H35NO6S and C26H21NO6S small molecules were, respectively, –46.0 and –41.9 kJ/mol, qualitatively agreeing with experiments. 34
Drugs based on mimetics that specifically target a single antiapoptotic protein can result in drug resistance. Conversely, drugs that act too broadly may damage healthy cells. An alternative is to design drugs based on mimetics that bind two or three anti-apoptotic molecules. Such a drug should have action broad enough to evade drug resistance, yet be specific enough to minimize damage to healthy tissue. The drug would likely be more cost-effective and cause fewer side effects than the use of multiple drugs, which each target an individual protein.
A promising drug candidate should be “ drug-like”, which means that it should have characteristics similar to known drugs. Bioavailability, protein affinity, toxicity, transport, absorption, and metabolic stability of a compound depend on many factors. These factors include the compound's hydrophobicity, electronic distribution, hydrogen bonding, and molecule size and flexibility.
The logarithm of octanol-water partition coefficient, logP, is recognized in drug design as a dependable gauge of the compound's hydrophobicity. High logP values are related to low absorption. A logP value smaller than 5 indicates that a compound is likely to be easily absorbed by the organism. 35 Another indicator of drug-likeness is the logS value (solubility). A reasonable value for logS is around –4, which translates to 0.1 mmol/liter. 35 Most commercially available drugs have logS values between –4 and –1..35,40 Molecular polar surface area (PSA) corresponds to the total surface area of polar atoms. PSA is a very valuable indicator of molecular transport through membranes. 36 PSA is used to predict a potential drug's absorption, including intestinal absorption, bioavailability, and blood-brain barrier penetration. A drug-like molecule should have a PSA value of less than 120 Å 2 . 36
Most commercially available drugs have molar masses of less than 500 g/mol. Molecules with higher masses are less likely to be absorbed and thus less likely to reach their target. The oral bioavailability of a drug can be predicted using the number of rotatable bonds, which measures molecular flexibility. 37 Oral bioavailability studies in rats (for over 1100 drug candidates) 37 have indicated that compounds with 10 or fewer rotatable bonds are highly likely to have good oral bioavailability. A relatively simple method that can help eliminate compounds that are not good drug candidates is Lipinski's Rule of Five. 38 The Rule states that most “drug-like” molecules have logP ≤ 5, molar mass ≤ 500 g/mol, 5 or fewer hydrogen bond donors, and 10 or fewer hydrogen bond acceptors. Molecules violating two or more of these rules may not be good drug candidates.
Our goal was to design new small molecule mimetics that would be drug-like and could potentially bind all three BCL-2, BCL-XL, and MCL-1 proteins, thus allowing apoptosis to proceed. The findings could facilitate the search for new drugs that are able to overcome the resistance of some cancers to conventional chemotherapy.
Methodology
Dataset
Three-dimensional (3D) experimentally known structures of the antiapoptotic BCL-2, BCL-XL, and MCL-1 proteins and the ABT-737 molecule were obtained from the Protein Data Bank (PDB), entries 2O22, 1PQ1, 2ROD, and 2YXJ, respectively. The structures of obatoclax (CID: 4905081) and ABT-702 (CID: 3965212) were obtained from ChemDB online databases. ChemDB was searched for small molecules that were similar in structure to obatoclax, ABT-737, or ABT-702. Experimental 3D structures of 47 relevant molecules were identified. Of these, 41 molecules were found to make either no stable poses or marginally stable poses with the BCL-2, BCL-XL, or MCL-1 proteins. The remaining six molecules formed stable poses with at least one of the proteins, while only three of these formed stable poses with all three proteins. These three small molecules, as well as ABT-737 and ABT-702, were used as templates to design new putative BH3 mimetics that formed stable complexes with all three BCL-2, BCL-XL, and MCL-1 proteins and possessed drug-like properties.
Computer Tools
The Deep View 39 and ArgusLab 30 computer programs were used to design the new putative mimetics and study their interactions with the BCL-2, BCL-XL, and MCL-1 proteins. ArgusLab is a docking program that facilitates molecular building and computational drug design. To perform docking one first needs to define atoms that make up the ligand and the binding site of the protein where the ligand should bind. During docking calculations the program attempts to position the ligand into the binding site and reports on any stable poses that it finds. ArgusLab can also perform semiempirical geometry optimizations.
Deep View is a user friendly program that allows simultaneous studies of several molecules. It greatly facilitates the design of molecular models. The molecules can be overlaid to reveal structural alignments and allow comparisons of their binding sites. Intuitive graphic and menu tools in Deep View allow easy mutations of amino acids and visualization of atomic bonds. The program can also perform energy minimizations. Lastly, good quality molecular images can be generated.
Two open-source programs, OSIRIS Property Explorer 40 and Molinspiration 41 were used to predict the drug-likeness of newly designed small molecules. The programs calculated the values of logP, logS, PSA, and molar mass; the numbers of rotatable bonds, H-bond acceptors, and H-bond donors; as well as the drug-likeness and overall drug scores. OSIRIS Property Explorer also estimated the risks of side effects, such as mutagenicity or tumorigenicity. To calculate the overall drug score, OSIRIS combined logP, logS, molar mass, drug-likeness, and toxicity risks in a single number to predict the molecule's overall drug potential.
Procedure Employed
The 3D structures of BCL-2, BCL-XL, and MCL-1 proteins were downloaded into the Deep View program. Amino acids within 6 Å of the proteins’ BH3 binding sites were selected for molecular building and docking studies in ArgusLab.
Experimentally known small molecules were docked in turn to BCL-2, BCL-XL, and MCL-1 proteins. Docking was done using the ArgusLab GA dock function, along with the flexible setting where the twisting of molecular bonds was allowed. The stability of each docked pose was evaluated using ArgusLab energy calculations. Three of the experimentally known molecules, CID 18177004, CID 11673546, and CID 11544167, formed stable complexes with all three proteins.
By systematically modifying and combining parts of the three molecules, ABT-702, and ABT-737, new small molecules were designed using the Molecule Builder function in ArgusLab. Atoms in the original molecules were systematically substituted or deleted. Some fragments were moved to a different site in the molecule or added from other molecules. The newly-obtained molecules were then docked in turn to BCL-2, BCL-XL, and MCL-1. The molecules with the highest affinities were further modified. This procedure identified five newly designed small molecules that bonded all three antiapoptotic proteins and had promising drug-like properties. The OSIRIS Property Explorer 40 and Molinspiration 41 were used to calculate drug-related properties for the five molecules as well as predict their drug-likenesses and potential side effects.
Results and Discussion
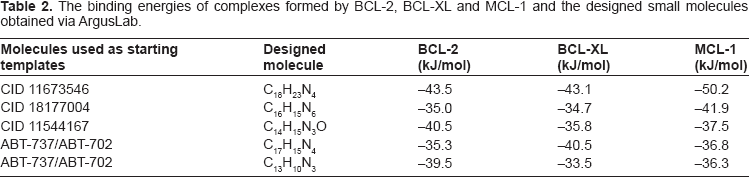
The binding energies of complexes formed by BCL-2, BCL-XL, and MCL-1 and the three small molecules originally identified in the ChemDB databases (CID 11673546, CID 18177004, and CID 11544167) are shown in Table 1. More negative binding energies correspond to higher binding affinities.
The binding energies of the stable poses formed by BCL-2, BCL-XL, and MCL-1 and the experimentally known small molecules obtained via ArgusLab.

Five new small molecules were designed from fragments of the three original molecules, ABT-737, and ABT-702, by substitution, insertion, and/or deletion, as described in the Methodology section of this paper. Figure 1 shows the structures of the five designed small molecules. The binding energies of complexes formed by the five designed molecules and BCL-2, BCL-XL and MCL-1 are given in Table 2.
The binding energies of complexes formed by BCL-2, BCL-XL and MCL-1 and the designed small molecules obtained via ArgusLab.

Structures of the five designed small molecules, from left to right: C18H23N4, C16H15N6, C14H15N3O, C17H15N4, and C13H10N3. Color code: C-yellow, N-pink, O-green, H-white.
Small molecule CID 11673546 was used as an initial template to design the C18H23N4 molecule. The new molecule had a high affinity for all three antiapoptotic proteins. As can be seen from Table 2, the energies of its stable complexes with BCL-2, BCL-XL, and MCL-1 were –43.5, –43.1, and –50.2 kJ/mol, respectively. These energies are, respectively, 12%, 10%, and 39% more negative than for the protein complexes formed with the unmodified CID 11673546 molecule.
The binding energies of BCL-2, BCL-XL, and MCL-1 complexes with the designed molecule C16H15N6 were –35.0, –34.7, and –41.9 kJ/mol, respectively (Table 2). C16H15N6 formed more stable complexes with BCL-XL and MCL-1 then the original molecule CID 11877004. Its binding energies with BCL-XL and MCL-1 became, respectively, about 1% and 6% more negative. Its binding energy with BCL-2 became less negative by about 11%. However, while the original molecule was not drug-like, the designed C16H15N6 molecule scored high on all studied drug-likeness criteria.
Complexes formed by the new molecule C14H15N3O and BCL-2, BCL-XL, and MCL-1 had binding energies of –40.5, –35.8, and –37.5 kJ/mol, respectively (Table 2). These energies were almost the same as for the original CID 11544167 molecule. Nevertheless, the designed C14H15N3O molecule was drug-like, whereas the original CID 11544167 was not.
Molecules C17H15N4 and C13H10N3 were designed from fragments of ABT-737 and ABT-702. Both molecules formed stable complexes with all three proteins. The binding energies resulting from docking C17H15N4 to BCL-2, BCL-XL and MCL-1 were –35.3, –40.5, and –36.8 kJ/mol, respectively (Table 2). The binding energies of the complexes between C13H10N3 and BCL-2, BCL-XL and MCL-1 were –39.5, –33.5, and –36.3 kJ/mol, respectively (Table 2). The formation of the stable complexes of these two designed molecules and MCL-1 is especially significant since the original ABT-737 does not interact with MCL-1. In addition, to the best of our knowledge, complexes of ABT-702 and BCL-2, BCL-XL and MCL-1 are not known.
Some general features could be observed for all five designed molecules. They all contain the pyrrole ring, which seems to play an important role in their binding. It is noteworthy that pyrrole is used to derive certain compounds that are utilized by pharmaceutical industries for drug production. The polar surface areas (PSAs) of all five molecules are between about 31 and 81 Å 2 , well below the “drug-likeness” cutoff of 120 Å 2 . In addition, their molar masses are less than about 303 g/mol. All of the five designed molecules have 6 or fewer rotatable bonds and H-bond acceptors, as well as 5 or fewer H-bond donors. All of these parameters have been shown to be reliable indicators of drug-likeness.35–37
Drug design entails devising small molecules that are complementary to the protein binding site. 42 In general, protein binding sites display specificity and affinity toward certain ligands. 43
When bound to the small molecules designed in this study, the BCL-2, BCL-XL and MCL-1 proteins experienced various degrees of structural changes when compared to their unbound states.
Day et al 42 showed that Noxa and Puma bonded to a hydrophobic groove created by MCL-1 residues M212, V230, V234, T247 and F251. In our study, we identified MCL-1 binding site which included the above residues. All five of the designed molecules bonded within this site. Figure 2 shows MCL-1 binding groove with designed molecule C17H15N4. The Noxa- and Puma-binding residues M212, V230, V234, T247, and F251 are indicated as yellow spheres in Figure 2.

The MCL-1 binding groove is shown with designed small molecule C17H15N4 (red ball-and-stick). MCL-1 residues that bind noxa and Puma, M212, V230, V234, T247, and F251, are shown as yellow solid spheres. Nearby MCL-1 residues are presented as purple solid spheres. To increase visibility of the small molecule and yellow residues, some MCL-1 residues are either presented as purple ball-and-stick or omitted.
Experiments showed that all BH3 ligands caused similar structural modifications of MCL-1 when bound to it. 42 In agreement with this, we found that changes in MCL-1 complexes were similar for all five designed molecules. Furthermore, a conformation of a bound MCL-1 was similar to that of the free MCL-1. The root mean square deviation, rmsd, between the free MCL-1 and the one bound to Noxa or Puma was measured previously to be about 1.4 Å. 42 Similarly, we found in Deep View that the rmsd between the free and bound MCL-1 was about 1.6 Å for all small molecule complexes.
Previous experiments indicated that the corresponding MCL-1 site was about 40 Å × 20 Å when binding Puma or Noxa. 42 Similarly, we found that the corresponding MCL-1 binding site was approximately 41 Å × 18 Å when binding the designed molecules. The major modification in bound MCL-1 in our studies is the 5% width increase of the binding pocket when compared to the free MCL-1. This result agrees with small changes found experimentally in MCL-1 complexes. 43
Even though their overall topology was preserved, BCL-2 and BCL-XL binding sites experienced greater modifications than MCL-1 site. The rmsd values found by Deep View between the free and bound forms of BCL-2 and BCL-XL were about 3.0 Å and 4.0 Å, respectively.
ABT-737 binds BCL-XL and BCL-2 within certain binding pockets. 44 We found that all five designed molecules were able to bind the same binding pockets as ABT-737. These results suggest that the designed molecules could potentially prevent interactions of BCL-2 and BCL-XL with proapoptotic BH3 proteins in a similar fashion as ABT-737 does.
Most traditional cytotoxic agents indirectly cause apoptosis by damaging DNA. 45 However, cancer cells often have damaged apoptotic pathways that allow them to avoid apoptosis. 46 Current strategies of cancer therapy target parts of the pathways that are essential for cancer survival. 47 The main goal is to sidestep damaged pathways and make the cancer cells susceptible to apoptosis.
Studies aimed at understanding how ABT-737 produces cytotoxicity in cancers 45 showed that ABT-737 stimulates apoptosis by specifically binding BCL-2 or BCL-XL proteins rather than by damaging the DNA..4,22 ABT-737 is effective in inducing apoptosis even at very high concentrations of its target proteins. ABT-737 was found to engage the same binding sites of antiapoptotic proteins as cellular BH3-only proteins. 22 Similarly, previous research showed that apoptosis stimulated by ABT-263 (an orally bioavailable agent similar to ABT-737) was the direct consequence of inhibition of BCL-2 family proteins. 48
Small molecules designed in this study were found to bind within the same binding groove as ABT-737. All of them share some of the structural characteristics of ABT-737. In light of previous studies on ABT-737, 22 the putative mimetics designed here would be expected to stimulate apoptosis in a similar fashion by specifically binding their target BCL-2 family proteins. If further experimental studies confirm this indeed to be the case, the potential of these new mimetics to fight cancer would be enhanced.
Acting either as a single agent or in combination with other chemotherapeutic agents, ABT-737 significantly increases the cytotoxic response in certain cancer cells. 49 Preclinical studies 22 show that the highly specific binding of ABT-737 reduces side effects, thus making it a promising candidate for clinical trials. Indeed, experimental studies showed that ABT-737 caused negligible undesired effects in mice. 4 Similarly, small molecule ABT-702 was shown to be orally active, highly target specific, and non-toxic in mice. 50
Two molecules designed in this study, C17H15N4 and C13H10N3, were made using ABT-737 and ABT-702 as starting templates. Similarly to these two ABT agents, the designed molecules showed affinity toward their target proteins. The binding propensities and limited cytotoxicities of ABT-737 and ABT-702, the oral activity of ABT-702, and predicted drug-likeness properties suggest that the designed molecules might likewise be promising candidates for clinical trials.
The “drug-likeness” of the small molecules designed here was estimated using OSIRIS Property Explorer 40 and Molinspiration. 41 OSIRIS Property Explorer reported that no side effects (such as mutagenicity or carcinogenicity) were likely for the designed molecules. Drug-relevant properties obtained by the two programs are given in Table 3. The number of rotatable bonds, H-bond acceptors, and H-bond donors were calculated by Molinspiration, while the Drug-likeness and Drug Score values were obtained by OSIRIS. The estimates from both programs for the molar masses and the logP and PSA values were similar. Table 3 reports the values found by OSIRIS Property Explorer.
Drug-like properties of designed putative small molecule mimetics estimated by Molinspiration and OSIRIS Property explorer. Relevance and acceptable values of the listed properties are described in the text. In the table, MM stands for the molar mass; nrotb for the number of rotatable bonds; nON for the number of H-bond acceptors; and nOHNH for the number of H-bond donors.
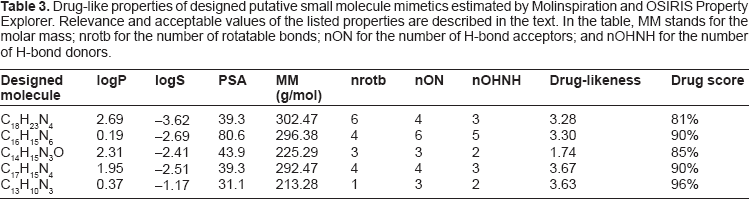
As can be seen from Table 3 all five of the designed molecules have logP ≤ 5, –5 ≤ logS ≤ 1, PSA less than 120 Å 2 , molar masses less than 500 g/mol, number of rotatable bonds fewer than 10, number of hydrogen bond donors 5 or fewer, and the number of hydrogen bond acceptors 10 or fewer. None of the designed molecules violate any of Lipinski's Rule of Five. They all have positive drug-likeness values with overall drug scores between 81% and 96%. All five molecules form stable complexes with MCL-1, BCL-2, and BCL-XL proteins. They also bind at the same sites as cellular BH3-only proteins, as well as at the same BCL-2/BCL-XL site where ABT-737 binds. These results and previous experimental and preclinical findings strongly suggest that the small molecules designed in this study may indeed be promising drug candidates. 35
Conclusions
Our research identified five new putative small molecule mimetics that could make stable complexes with three antiapoptotic BCL-2, BCL-XL, and MCL-1 proteins. These results are novel because, to our knowledge, there are not many, if any, small molecules known to bind all three BCL-2, BCL-XL, and MCL-1 proteins.
The new small molecules were obtained by computationally modifying experimentally known molecules. The binding energies of complexes formed by the designed molecules and BCL-2, BCL-XL, and MCL-1 proteins ranged between –33.5 and –50.2 kJ/mol. Especially significant is the formation of a stable complex of modified ABT-737 with MCL-1 since the original ABT-737 does not interact with MCL-1. The modified ABT-702 also formed stable complexes with BCL-2, BCL-XL, and MCL-1. It is noteworthy that, to the best of our knowledge, stable complexes of ABT-702 and BCL-2 family proteins have not been studied previously. Drug-likeness studies performed on the five designed molecules, as well as previous experimental and preclinical studies on ABT agents and BH3-only cellular proteins, strongly suggest that the putative new mimetics may indeed be promising drug candidates.
Our findings present both possibilities and challenges for anticancer drug design. Drugs based on the small molecules studied here might be useful against cancers that overexpress BCL-2, BCL-XL, and/or MCL-1 proteins. A single drug based on these molecules should cost less and cause fewer side effects than a combination of drugs each aimed at a single antiapoptotic protein. Molecules designed in this work formed strong bonds with BCL-2, BCL-XL, and MCL-1 in silico. However, these molecules may not be easy to synthesize in a lab. Also, once synthesized, they might not bind to their anticipated target proteins in living cells.
The usefulness of the designed molecules for anticancer drug development will eventually be determined by their abilities to decrease BCL-2, BCL-XL, and MCL-1 expressions in living cells and to selectively target cancerous versus healthy cells.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.