
Abstract
We present a novel machine learning approach for the classification of cancer samples using expression data. We refer to the method as “decision trunks,” since it is loosely based on decision trees, but contains several modifications designed to achieve an algorithm that: (1) produces smaller and more easily interpretable classifiers than decision trees; (2) is more robust in varying application scenarios; and (3) achieves higher classification accuracy. The decision trunk algorithm has been implemented and tested on 26 classification tasks, covering a wide range of cancer forms, experimental methods, and classification scenarios. This comprehensive evaluation indicates that the proposed algorithm performs at least as well as the current state of the art algorithms in terms of accuracy, while producing classifiers that include on average only 2–3 markers. We suggest that the resulting decision trunks have clear advantages over other classifiers due to their transparency, interpretability, and their correspondence with human decision-making and clinical testing practices.
Introduction
Numerous studies have been conducted to investigate if high-throughput gene and microRNA (miRNA) expression datasets can be used to differentiate between normal and tumor tissue, between tumors from different cancer forms, tumors in different stages, different responses to therapy, and so on. The earliest studies (eg, by Alon et al 1 and Golub et al) 2 focused on the use of unsupervised clustering analysis, whereas subsequent work exhaustively evaluated the usefulness of supervised classification approaches using standard machine learning methods.3–8 A challenge in this scenario is to produce a classifier that is accurate and robust, while at the same time being interpretable, transparent, and able to provide some biological insight into the classification process.
Machine learning methods that have been applied to transcriptome data include “black box” methods such as support vector machines (SVM)3,4,8 and artificial neural networks (ANN),9–11 as well as symbolic and rule-based methods, such as decision trees.11–13 The black box methods are statistically powerful and often provide good classification accuracy, but suffer from the drawback of producing non-transparent classifiers, which offer little or no insight into the basis for the classifications. In addition, these methods rely on using the complete marker set, or a large subset thereof. Decision tree methods, on the other hand, provide transparent and easily interpretable classifiers based on a relatively small set of markers, but suffer from the drawbacks of over-fitting, sensitivity to noise, and poor generality. Consequently, decision tree algorithms such as J48 and C4.5 have been among the worst performers in several comparisons of machine learning classification algorithms across different sets of cancer-related expression data.14–17 The weaknesses of the decision tree approach can be counteracted by using random forests; 18 however, this has the drawback of introducing complexity and producing models where the classification process is so non-transparent that it is essentially a black-box classifier.
A few algorithms have been proposed in efforts to overcome these challenges. A notable example is the top-scoring pairs (TSP) algorithm, which was introduced by Geman et al.
15
The central idea of the TSP algorithm is to identify pairs of markers showing contrasting expression levels. If marker
An extended variant of TSP, called
Other approaches have been proposed to reduce the number of markers used for classification and/ or to make the classifier more interpretable. Lauss and colleagues proposed a method that uses the receiver operating characteristic (ROC) to choose a small subset of genes and then builds a classifier on “metagenes,” which are expression profiles corresponding to the average expression values of the chosen genes across the set of probes. 20 Another variation on the top-scoring pairs theme has also been proposed where doublets were formed by different functions, such as the vector sum or difference between expression vectors of gene pairs. 17 These doublets were then used as input into five standard classification methods, including SVM and decision trees, and improved accuracy was observed in comparison to the standard approach of using expression values of individual genes as input.
Herein we present a new classification algorithm, called the Decision Trunk Classifier, which has many features in common with decision trees, but with adaptations designed to make the algorithm more suitable for building classifiers for transcriptome data in clinical application scenarios. The goal of these adaptations is to gain classifiers that are smaller, simpler, and easier to interpret than standard decision trees, while at the same time achieving higher classification accuracy. An additional goal is to have a minimum number of parameters, since this will facilitate algorithm use, and should make it possible to avoid the over-fitting and inflated estimates of performance which may result from evaluating many combinations of parameter settings.
The key idea behind decision trunks is that for each internal node (marker) added to the “trunk,” all remaining samples are divided into three groups, for example a set of samples where the marker gene has: (1) high expression, (2) low expression, and (3) medium expression. To this node, three outgoing branches and nodes are then added, with one corresponding to each group, and with the “low” and “high” nodes being leaf nodes where a decision is made according to the majority class of the group. The third, “middle” node corresponds to uncertain cases and is therefore an internal node where no decision is made. Instead, a new marker is chosen for this node, and the reduced set of samples is again divided into those with low, high, and intermediate expression levels for this marker. This process continues until a stopping criterion is reached and a final marker is chosen. The node for this marker has only two outgoing branches (for low and high expression), both leading to leaf nodes. The result of this process is a “slim” decision tree, consisting only of a straight “trunk” with decision nodes pointing out from the trunk.
We hypothesize that the use of a medium expression level interval for uncertain cases, and deferring the decision to the next level of the trunk, will result in higher robustness to noise than in standard decision trees. We also claim that decision trunks will be easier to interpret and relate to the underlying biology, as well as to clinical testing practices, than standard decision trees. The proposed method has been implemented and tested on a large number of expression datasets, and its classification accuracy has been compared to a wide variety of other methods, including both standard machine learning algorithms and special-purpose algorithms designed for expression data.
Methods
Algorithm Overview
As input, the decision trunk classifier requires a dataset consisting of expression values for
Usage of the algorithm can be divided into three steps: (1) building decision trunks; (2) choosing the number of decision levels; and (3) evaluating classification accuracy. During the first step, five sets of decision trunks are built with
Building Decision Trunks
To build a decision trunk, the algorithm first creates a decision trunk object and then adds to it one decision level at a time. For every level, a

where

When the decision thresholds have been set, the algorithm removes all samples whose expression values are lower than
Figure 1 illustrates the process of generating decision nodes. The samples are ranked according to their expression values for the most significant marker and the upper and lower thresholds for expression value
Choosing the Number of Decision Levels
To choose the number of decision levels, the algorithm checks how many features are selected at each level when decision trunks are built from the
Feature Selection
In addition to the

We refer to this method as maximum class polarization (MCP), as it selects the feature that results in the most polarized division of the samples into two groups. Similar feature selection methods have been suggested and tested by Park et al 21 and Dettling and Bühlmann, 12 and these methods can be considered as adaptations of the Wilcoxon test statistic.
Implementation
The main part of the decision trunk algorithm was implemented in Perl. For efficiency reasons, the feature selection methods were implemented as a shared C library. The program is executed from the command line and requires a text file containing an expression data table as input. The table must have column names on the first row and row names in the first column. Furthermore, the user is expected to supply the name of the classification variable to be used. Class information is supplied as metadata (rows starting with “#”) at the top of the file. To facilitate the generation of the metadata, the program also accepts a supplementary file with the -s argument. This file should have a header, followed by any number of rows. These rows start with a sample name, followed by columns containing class labels. In the header of these columns, the names of the class variables should be given. More details on the usage of the software are supplied in the online documentation. The implementation of the algorithm is available as the Algorithm::TrunkClassifier package at CPAN.
As output, the program reports: (1) the accuracy for each round of evaluation (LOOCV or split-sample), as well as the overall average accuracy; (2) the decision trunk classifiers generated during the evaluation; (3) to which class and at what level in the decision trunk each sample was classified; (4) a log file containing the parameters used and sizes (number of samples) of the two classes.
Datasets and Evaluation Methods
The datasets used in this study contain expression data for a set of features (genes, miRNAs or NGS reads) from 13 published studies covering cancers of the prostate, bladder, breast, and lung, as well as neuroblastomas. The majority of studies were used for more than one classification task. For example both normal versus malignant and early versus late stage were used for the Sanchez bladder cancer data. Thereby, a total of 26 different classification tasks were formulated. A summary of the datasets and classification variables is given in Table 1. For bladder cancer datasets, the early stage was defined as Ta/T1, and late stage as ≥ T2. For breast cancer datasets, histologic grade 1 was considered as low grade and histologic grade > 1 as high grade. For neuroblastoma datasets, the International Neuroblastoma Staging System (INSS) stage 1–2 was defined as early stage and INSS stage > 2 as late stage. The Sanchez, Stransky, WangY, Sotiriou and Janoueix datasets were log2-transformed before classification. All datasets were examined for missing values and genes with >5% missing values were removed. The remaining missing values were imputed by taking the average of all values for that gene.
Description of datasets and classification variables.

NM, normal versus malignant; ST, stage; TR, treatment status; GR, grade; ER, estrogen receptor status; M Y, MYC amplification status; HI, histological subtype; DI, differentiation status; SU, survival.
The evaluation of classification accuracy was performed using LOOCV with the number of folds equal to the number of samples. The average accuracy for the given dataset was defined as the proportion of correctly classified test samples (which is equivalent to the number of true positives plus the number of true negatives divided by the total number of samples). The performance of the decision trunk algorithm was compared with that of the following algorithms: Naïve Bayes (NB), Decision Trees (J48), Voting Features Interval (VFI), SVM, single layer Artificial Neural Network (ANN), k-Nearest Neighbor (kNN), Prediction Analysis for Microarrays (PAM), TSP, and the Metagene Classifier (ROCC). Weka version 3.6.6 was used for NB, J48, VFI, SVM, and ANN, whereas Bioconductor packages were used for kNN (ver. 7.3–1), PAM (ver. 1.54), TSP (version 2.8), and ROCC (version 1.2). All algorithms were run with default parameters except kNN and PAM. For kNN, the parameter
In addition to cross-validation, the decision trunk algorithm was further evaluated using three different approaches. The first approach consisted of a split-sample procedure, where each one of the 26 datasets was divided randomly ten times into a training set, containing 80% of the samples, and a test set containing the remaining samples. In the second approach 40 artificial datasets were generated using two random normal distributed variables, one for each class. Each dataset consisted of 200 samples per class, with the two classes having equal standard deviations but different means. The standard deviations were set to 0.5, 1.0, 1.5, 2.0 (four values) and the difference between the means was set to 0.5, 1.0, …, 5.0 (ten values). The third approach aimed to test the generality of the algorithm by using data from one study as the training set and data from another study as the test set. This was done using the Sanchez dataset for training and the Stransky data for testing, and vice versa. This combination of datasets was chosen since both studies were on bladder cancer and aimed to address the same classification problems, namely grade and stage.
Results
The decision trunks generated for the 26 datasets had on average 2.0 levels. To illustrate the argument that this gives transparent and easily interpretable classifiers, Figure 2 shows the decision trunk for the Carlsson1 dataset (quantitative polymerase chain reaction (qPCR) data on prostate cancer tumors versus healthy prostate tissue). Only two markers, miR-126* and miR-892b, are included in the classifier. Samples with very low qPCR values for miR-126* (<23.2) are assigned class N (normal) and samples with very high values for miR-126* (>25.28) are assigned class M (malignant). Samples with intermediate values for miR-126* are assigned a class based on their expression of miR-892b. LOOCV accuracy for this simple classifier was 97.36%. The accuracy of classifiers generated by the other algorithms on the same dataset ranged from 76.32% (SVM) to 94.47% (NB, J48, VFI, PAM, and TSP). The microRNA miR-126* has been identified in many studies as being associated with several cancer forms, including prostate cancer,22,23 while miR-892b has not previously been recognized as deregulated in cancer.
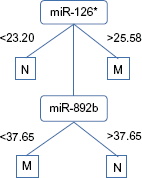
Example of decision trunk generated from the Carlsson1 dataset.
Classification accuracies on all datasets of the decision trunk classification algorithm (DTC), as well as all the algorithms evaluated for comparison, are presented in Table 2. A general trend is that the average performance of all algorithms is quite high. DTC had the highest average classification accuracy (84.47%) and was the best performing of all algorithms on 11 of the 26 datasets (42%). The second and third highest average classification accuracies were achieved by ROCC (83.71%) and kNN (81.34%), which where the best performing algorithms on five (19%) and one (4%) datasets, respectively. ANN also reached > 80% in average classification accuracy, while the remaining algorithms (NB, J48, VFI, SVM, PAM, and TSP) had average accuracies in the range 70% to 80%. It is noteworthy that three of the five best performing algorithms in this evaluation (DTC, ROCC, and TSP) have been designed for expression data, and have the goal of using a minimal number of markers. General-purpose classification algorithms, such as NB, J48, VFI, and SVM, as well as special purpose algorithms using a large set of markers (ie, PAM), generally gave poorer results.
Classification accuracies for all algorithms as determined by leave-one-out cross validation.

Comparing the decision trunk classifier to the standard decision tree algorithm J48 reveals that DTC seems to do better on average by being more robust. The two algorithms show comparable performance for many datasets, but J48 occasionally fails and performs considerably worse. This occurred in particular on the datasets Sanchez_GR (87.91% versus 59.34%), Takeuchi_SU (68.45% versus 41.61%), Stransky_ST (75.43% versus 52.63%), and Takeuchi_DI (86.44% versus 66.10%). The higher robustness of DTC can also be seen in the standard deviations of the two algorithms' accuracies over all datasets, which are 9.6 and 16.0, respectively. A one-sided Student's

Relationship between classification accuracy and the number of decision nodes (markers) for J48 decision trees (red) and the decision trunk algorithm (black) on the 26 datasets.
The accuracies for the DTC reported in Table 2 were obtained when using the stability criterion (see Methods) for selection of the number of levels (
Decision trunk accuracies achieved using a posteriori selection of optimal number of levels (left) and using stability criteria (right).

The split-sample approach was applied to further evaluate the robustness of the DTC algorithm. Each of the 26 datasets was repeatedly split into training and test sets and the average classification accuracy recorded (Supplementary Table 1). The resulting average accuracy sank to 79.17%. Thus, after this moderate drop of just over five percentage points, the average accuracy of the DTC was still above the performance of J48 and four other algorithms in the cross-validation (compare with Table 2).
The difficulty of a classification task depends on the distributions of expression values for the two classes to be separated. The greater the overlap between the distributions, the more difficult it will be to classify the samples correctly. To investigate this relationship between distribution and accuracy, 40 artificial datasets with a range of standard deviations and differences between means were generated. The average accuracies of decision trunks generated using these datasets are shown in Figure 4, along with the accuracies for the cancer datasets. Accuracies range from 80%–100% for artificial datasets with an SD = 0.5, regardless of the difference between means, while 1 ≤ SD ≤ 2 requires a difference between means of 1 to 2.5 to achieve 80% accuracy. For comparison, the accuracies from the 26 cancer classification tasks (Table 2) were also plotted in Figure 4. Considering the SD for the top-level marker in the trunks generated from these datasets, most of the classification accuracies are similar to those obtained from the artificial data. There are cases to the far right in the plot, where classification accuracy on cancer datasets is lower than expected. However, these datasets had very high SD (up to 9.6), which explains why accuracy is lower than on the artificial datasets where SD was limited to a maximum of 2.

Decision trunk classification accuracies for artificial and cancer datasets.
As a test of generality, the DTC algorithm was applied using data from one study as the training set and data from another study as the test set. The two microarray gene expression datasets (Sanchez 31 and Stransky) 32 concern bladder cancer and contain a stratification of tumors according to grade and stage. The probe IDs in both datasets were converted to HUGO Gene Nomenclature Committee symbols, and genes unique to either dataset were removed. Normalization was carried out by dividing the expression values for each gene by the mean expression of the gene, for each dataset separately. Four classification tasks were then carried out, namely by training on Sanchez, and classifying Stransky (and vice versa), first for tumor grade and then for stage. Comparing the results with Table 2, it can be seen that there is essentially no loss of accuracy when classifying the Stransky grade data: 87.27% when training on the Sanchez data versus 89.09% when training on the Stransky data. When classifying the Stransky stage data, accuracy dropped to 66.67% when training on the Sanchez data, compared to 75.43% when training on the Stransky data. Loss of accuracy was generally greater when classifying samples from the Sanchez dataset: from 87.91% to 65.93% for grade and from 90.11% to 73.63% for stage. The greater loss in accuracy when training on the Stransky data can be explained by the fact that the Stransky dataset contains 55 (grade) and 57 (stage) samples, while the Sanchez dataset contains 91 samples for both grade and stage.
To further evaluate the usefulness of selected genes as cancer biomarkers, a comparison of selected markers was conducted between the DTC algorithm and J48. The idea is that if several different approaches identify a gene as a useful marker for classification, it is more likely to be robust. For DTC and J48, the root nodes were compared to determine the overlap of marker selection. Only in six of the 26 classification tasks did the algorithms choose the same marker for their top-level nodes, and this corresponds to the cases where both algorithms perform well.
Discussion
Comparison with other Decision Tree Variants
There are several variations on the theme of decision trees. A simplified version, named decision stumps, generates classifiers containing a single decision node with
Decision trunks bear some resemblance to fuzzy decision trees, 25 a method designed to address the problem that decision trees were originally designed for attributes that take on a discrete set of values. For continuous domains, such as expression data, the attributes must be discretized. This can be done by partitioning the attribute range into two intervals, 26 but the “crisp” cutting points resulting from standard decision trees produce high error rates in many real-world applications due to vague and noisy data. 27 Fuzzy decision trees were designed to overcome this problem by implementing a “soft” form of discretization. 25 A standard decision tree, using crisp discretization, partitions the decision space into a set of non-overlapping subspaces and assigns each object a particular class. In contrast, a fuzzy decision tree, using soft discretization, allows an object to be associated with different paths in the tree and assigns the object a probability of belonging to each class. 27 This has been shown to improve robustness in many applications, but at the cost of a more complex training procedure, the introduction of parameters, and trees that are not as easy to interpret as standard decision trees. Decision trunks offer a third alternative, which can be seen as containing some elements of both approaches. The decision trunks algorithm sets crisp thresholds, but only to identify those samples that are clearly defined by a given probe, while deferring decisions on the remaining samples to the next lower level in the trunk. The results presented in this paper indicate that this improves generality and robustness, while giving classifiers that are even simpler and easier to interpret than standard decision trees.
The limited performance of standard decision trees on expression data has also been addressed by using boosting.3,7,12 The improved classification accuracy of such approaches comes with drawbacks such as the introduction of additional parameters (eg, the number of boosting iterations), and an increased computational cost.
Alternative Methods for Feature Selection
The standard method for feature selection in decision tree algorithms is information gain. Information gain is based on the entropy function,
In order to facilitate comparisons with standard decision tree algorithms, we would want to use the information gain criterion for the selection of markers in decision trunks. This would give a clearer idea of what feature(s) of the decision trunks algorithm that the gains in classification accuracy can be attributed to. Unfortunately, the information gain criterion cannot be implemented in decision trunks without modifications. The decision trunks algorithm adds three outgoing branches from each decision node (in a binary classification problem), and the daughter nodes have different interpretations. The two leaf nodes are, by definition, maximally pure and the middle node is, by definition, impure. Since the upper and lower decision thresholds are calibrated by the quartiles of the sample set, the reduction in impurity (ie, information gain) is constrained in such a way that a huge number of marker and threshold combinations would achieve the same information gain. Thus, information gain is not a meaningful marker selection criterion for classifiers of this type.
Further Development
An attractive feature of decision trees, which also applies to decision trunks, is that a set of symbolic rules can be derived from a decision tree and implemented in a rule-based decision system.
14
The decision nodes on the path leading to the leaf node generate a conjunctive antecedent and the classification specified by the leaf generates the consequent of an “if-then” rule on the form
It can be advantageous in some application scenarios to be able to build classifiers with the option to assign the class label “uncertain” to some samples.
12
This could easily be achieved by a slight modification of the decision trunks algorithm, by simply letting the bottom-level node of the decision trunk have three outgoing branches, labeled
The decision trunks algorithm is designed for binary classification problems. Multiclass problems can nevertheless be handled using the one-against-all approach, which is commonly used in the machine learning community.28,29 It reduces a classification problem with
Conclusions
The decision trunk algorithm provides classifiers that: (1) involve a minimal set of markers; (2) are as accurate as classifiers produced by the most powerful machine learning methods; and (3) are easily interpretable and correspond to clinical test procedures. In addition, the algorithm is essentially parameter-free since it is possible to use the stability criterion for setting the parameter
Author Contributions
Conceived the idea of a decision trunks algorithm: BO. Jointly developed algorithm details: BU, BO. Implemented the algorithm: BU. Jointly designed the evaluation experiments: BU, BO. Collected the datasets and carried out all experiments: BU. Jointly analyzed the results: BU, KKL, BO. Developed the structure and main arguments of the paper: BO. Made critical revisions and updates of the manuscript: BU, BO, KKL. All authors reviewed and approved of the final manuscript.
Funding
This work was partially funded by the Swedish Knowledge Foundation, (grant 2009/0291).
Competing Interests
Author(s) disclose no potential conficts of interest.
Disclosures and Ethics
As a requirement of the publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy, and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.