
Abstract
In this study, we use a well-defined mouse model to examine tissue factor's (TF) role in osteolytic bone metastasis. C57BL/6 mice received either mock siRNA-transfected or TF-specific siRNA-transfected B16F10 melanoma cells by left ventricular injection. A third group served as an age-matched control and did not receive any tumour cells. The effect on tumour burden and bone strength was then determined 14 days later by using bone histomorphometry and biomechanical testing. Based on histomorphometric analysis of the femurs, mice receiving TF-specific siRNA-transfected tumour cells had significantly reduced tumour burden as compared to those from mice that received mock siRNA-transfected tumour cells (2.20 ± 0.58% vs. 9.18 ± 2.20%). Furthermore, the femurs from mice receiving TF siRNA-transfected tumour cells displayed decreased osteoclast surface and consequently, increased cancellous bone volume and strength when compared to those isolated from mice that were injected with mock-transfected tumour cells. More importantly, no differences in osteoclast surface or cancellous bone volume and strength were observed when the femurs of mice that received TF siRNA-transfected tumour cells were compared to control mice that did not receive tumour cells. Based on these findings, we conclude that the expression of TF by tumour cells promotes their ability to metastasize to bone, thereby facilitating tumour cell—induced cancellous bone loss.
Introduction
Bone is the third most common site to which solid tumours metastasize. 1 Cancers, particularly melanoma and carcinomas of the breast, prostate, and lung frequently metastasize to bone. The metastasis of tumour cells to bone is a devastating complication of cancer. It is often associated with extensive osteolysis which can cause significant bone destruction and result in hypercalcaemia.2–4
While the metastasis of cancer to bone is a major cause of morbidity and mortality, little is known about the mechanism by which it occurs. Whether factors that play a role in the hematogenous spread of cancer to organs such as lung, also play a role in the metastatic spread of cancer to bone is unknown. For example, TF is a 47-kDa transmembrane glycoprotein that acts primarily as a cellular initiator of coagulation by binding to factor VII (FVII).5–7 It is expressed by smooth muscle cells, fibroblasts, and stimulated endothelial cells as well as numerous cancers, including melanoma and cancers of the breast, prostate, pancreas, lung, stomach and colon. 8 Studies have correlated the metastatic ability of tumour cells to TF expression found either on the tumour cell surface or on cell membrane-derived microparticles shed from the tumour cells.9–13 However, while previous studies have implicated TF in pulmonary metastasis, to date, no study has demonstrated a role for TF in the process of cancer metastasis to bone.
How the expression of TF by tumour cells acts to increase their metastatic potential is unclear. Based on models of pulmonary metastasis, TF expression by cancer cells has been postulated to increase thrombin generation and to thus, increase fibrin deposition on the tumour cell surface. This in turn is thought to lead to the formation of tumour cell/platelet emboli, which served to protect the cancer cell from host immune responses. 14 Alternatively, thrombin has been proposed to increase the expression of specific adhesive moieties on either the tumour cell or endothelial cell surface, thus promoting tumour cell adhesion and eventually, extravasation.15,16
In the present study, we use a well-defined murine model of osteolytic bone metastasis to determine if TF expression by cancer cells influences their ability to metastasize to bone. Specifically, we inhibited TF expression in B16F10 murine melanoma cells by transfecting them with a TF-specific short interfering RNA (siRNA) prior to administering the tumour cells by left ventricular injection to mice. We then used bone histomorphometry and biomechanical testing to determine how this affected the metastatic process. Herein, we demonstrate that a transient reduction of TF expression in murine melanoma cells inhibits their ability to metastasize to bone, and thus, prevents tumour cell—induced pathological changes from occurring.
Materials and Methods
Materials
Pathogen-free female C57BL/6 mice age 10–12 weeks old were purchased from Charles River Laboratories (St. Constant, QC, Canada). The td- tomato expressing vector was a gift from Dr. John Lewis, University of Western Ontario, (London, ON, Canada). Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), penicillin, streptomycin, hygromycin B, Lipofectamine 2000 as well as the mock and TF siRNA sequences were purchased from Invitrogen (Burlington, ON, Canada). The JB-4 Embedding Kit was obtained from Polysciences, Inc. (Warrington, PA, USA). A tartrate-resistant acid phosphatase (TRAP) kit and acid hematoxylin solution were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Transfection and selection of a stable td-tomato B16F10 murine melanoma cell line
B16F10 melanoma cells were transfected with an expression vector containing the coding sequence for td-tomato fluorescent marker using Lipofectamine 2000 according to the manufacturer's specifications. Briefly, B16F10 melanoma cells were plated into 100 mm culture dishes containing DMEM supplemented with 10% FBS. When the culture reached approximately 70% confluency, the cells were transfected with 12 μg of the td-tomato expressing vector. Seventy-two hrs later, cells were trypsinized and subcultured 1:20 into 100 mm culture dishes in complete media supplemented with 500 μg/ml hygromycin B. Fluorescent colonies were identified using a Leica DMIL microscope and propagated in complete media containing 500 μg/ml hygromycin B at 37 °C.
Transfection of td-tomato-expressing B16F10 murine melanoma cells with TF-specific siRNA
Td-tomato—expressing B16F10 melanoma cells were transfected with either mock siRNA or TF-specific siRNA using Lipofectamine reagent. Briefly, 6 × 105 td-expressing B16F10 melanoma cells were seeded into 6 cm dishes. Twenty-four hrs later, the cells were transfected with 100 nmol/L of either TF-specific (5′-GCAUUCCA GAGAAAGCGUUUA-3′ (sense), 5′-AACGCUUU CUCUGGAAUGCCU-3′ (antisense)) or mock siRNA (5′-GCGCUUCAGGCA CUACAAAUA-3′ (sense), 5′-UUUGUAGUGCCUGAAGCGCCG-3′ (antisense)). 11 Approximately 24 hrs following transfection, the cells were harvested and administered to C57BL/6 mice by left ventricular injection.
Semi-quantitative reverse transcriptase—polymerase chain reaction (RT-PCR)
Total RNA was isolated from either untransfected, mock-transfected, or TF-specific siRNA-transfected B16F10 melanoma cells using the RNeasy mini kit (Qiagen, Sciences, Maryland, USA). cDNA was reverse transcribed from 4 μg of total RNA using Superscript II Reverse Transcriptase (Invitrogen, Burlington, ON, Canada). RT-PCR analysis of the cDNA was carried out using PCR Supermix (Invitrogen, Burlington, ON, Canada), and the forward and reverse TF primers: 5′-GCAGGCATTCCAGA GAAAGCG-3′ and 5′-TCTCCCAGGAAACTCTTCCATTG-3′, respectively. The reverse primer was labeled at the 3′end with 32 P using polynucleotide T4 kinase (Invitrogen, Burlington, ON, Canada). Amplification of the PCR products was performed using a PTC-100 Programmable Thermal Controller (MJ Research Inc., NV). Twenty-six cycles and an annealing temperature of 60 °C were determined to be the most appropriate conditions for RT-PCR (data not shown). PCR products were electrophoresed on a 7.5% polyacrylamide gel in triplicate, exposed to Kodak BioMax XAR film overnight and quantified using ImageQuant Software, Version 5.2 (Molecular Dynamics).
Clotting assay
A clotting assay was used to measure TF activity of B16F10 cells in vitro. Briefly, 20 μl of either control, mock siRNA-transfected or TF siRNA-transfected B16F10 cells (1.12 × 104 cells/μl) were incubated with 100 μl of a plasma mixture (9:1 ratio of human to mouse plasma) for 5 min at 37 °C. After the incubation, 20 μM of CaCl2 was added and turbidity measured over time at 350 nm using a SPECTRAmax plate reader (Molecular Devices, Sunnyvale, CA). Clotting times for each condition were recorded. All conditions were carried out in duplicate.
Experimental design
A total of 21 C57BL/6 female mice were randomized into one of three treatment groups, each consisting of seven animals. Two groups received either a total of 5 × 105 TF siRNA-transfected B16F10 cells or an equivalent number of mock-transfected cells by left ventricular injection. A third group served as an age-matched control and did not receive any tumour cells. All animals were sacrificed on day 14 by CO2 asphyxiation and after exsanguination, the femurs were removed for histomorphometric analysis.
In a second study, C57BL/6 female mice were again randomized into one of three treatment groups (n = 6 mice/group). Two groups received left ventricular injections of either TF-specific siRNA-transfected tumour cells or mock siRNA-transfected tumour cells at a concentration of 5 × 105 cells/animal. The third group did not receive tumour cells and served as an age-matched control. On day 14, all animals were sacrificed, and after exsanguination, their femurs were removed and immediately subjected to biomechanical testing.
Bone histomorphometry
Bone histomorphometry was performed as described previously. 17 Briefly, undecalcified femurs were fixed in 5% paraformaldehyde solution and then embedded in glycomethacrylate (JB-4 embedding medium; Polysciences, Inc, Warrington, PA, USA) as per manufacturer's instructions. Histological sections (6–8 μm) were made from the embedded femurs using a Leica RM2125RT microtome fitted with a Reichert-Jung 16 cm/d blade. Sections were mounted onto glass slides, air dried for 24 hrs, and either cover-slipped immediately or stained with tartrate-resistant acid phosphatase (TRAP) prior to being cover-slipped. A region that extended 1800 μm below the epiphyseal growth plate and included the entire metaphysis was viewed under either a light or fluorescent microscope using a Mertz grid (Carl Zeiss, Don Mills, ON, Canada). Parameters analysed included tumour burden, cancellous bone volume, trabecular number, width and separation, as well as osteoclast surface. For each section, tumour burden and cancellous bone volume were calculated from over 4000 point measurements using the Mertz grid at a magnification of 400×. Tumour burden was determined by viewing bone sections under fluorescent microscopy and identifying tumour cells by cellular fluorescence, while cancellous bone volume and trabecular parameters were analyzed directly under light microscopy. To determine osteoclast surface, bone sections were viewed under oil immersion (1000×) using light microscopy, and osteoclasts identified as large, multi-nucleated cells that stained red for TRAP activity. The histomorphometric parameters of trabecular width, separation and number were determined directly by using a fluorescent microscope (Zeiss Axioscope 2, Carl Zeiss, Don Mills, ON, Canada) coupled to an IBM computer. All images were taken using a 3CCD camera module and analyzed using the Northern Eclipse imaging software system (Empix Imaging Inc., Mississauga, ON, Canada).
Faxitron X-ray analysis
To visualize areas of osteolysis, X-ray images were taken on day 14 of the distal ends of femurs from mice in each condition (control, mock-transfected or TF siRNA-transfected) using a Faxitron cabinet X-ray system (model #43855A; Faxitron X-ray Corporation, Wheeling IL, USA).
Biomechanical testing
Biomechanical parameters of bone strength were measured for each femur using an Instron Biomechanical Testing System (Instron 442 loading frame; Instron, Burlington, ON, Canada). Briefly, femurs were placed on two lower supports (8 mm apart) with their anterior side facing upward. The femurs were then loaded at a rate of 10 mm/min by a compressive force from above until a fracture occurred at the mid-diaphysis. Biomechanical parameters such as force at maximum load, stress at maximum load, stiffness, elastic modulus, energy to break and toughness were calculated from the generated force-displacement and stress-strain curves using the Merlin series IX automated materials testing software, version 7.51.
Statistics
Analysis of variance was used to compare the data in the experimental group with those of the controls. An unpaired Student t-test with a Bonferroni correction for multiple comparisons was used to compare the significance of values between groups. P values < 0.05 were considered statistically significant. All data are expressed as a mean ± the standard error (SEM).
Results
Tumour burden in mice injected with TF-specific siRNA- transfected tumour cells
To determine if TF plays a critical role in the ability of tumour cells to metastasize to bone, we first transfected B16F10 melanoma cells with TF-specific siRNA, and then determined the effect on TF expression. As shown in Figure 1A, TF siRNA-transfected tumour cells demonstrated a significant reduction in TF mRNA expression at 24 hours when compared to either mock-transfected or untransfected tumour cells (81.2 ± 5.2% and 80.4 ± 6.8%, respectively; P < 0.005). This reduction in TF mRNA expression was transient, gradually returning to normal within 5 days (Fig. 1B). To ensure that the observed decrease in TF mRNA expression correlated with a reduction in TF activity, a clotting assay was also performed. As expected, the TF siRNA-transfected tumour cells took substantially longer to clot than both the untransfected tumour cells (875 ± 155 sec vs. 357 ± 32 sec, P < 0.05) and mock-transfected tumour cells (875 ± 155 sec vs. 362 ± 33 sec, P < 0.05) (Fig. 1C).
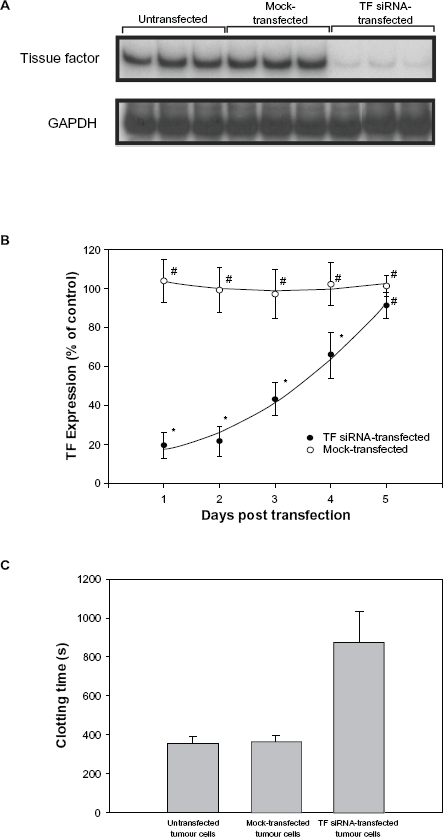
The effect of TF-specific siRNA on TF mRNA expression and activity. B16F10 murine melanoma cells were transfected with either mock siRNA or TF-specific siRNA, or were left untransfected. Total RNA was then prepared and analysed for the expression of TF mRNA using semi-quantitative RT-PCR as described in the Materials and Methods (Panels A and B). Alternatively, untransfected and transfected B16F10 melanoma cells were harvested and incubated with a mixture of plasma and CaCl2 to determine clotting times (Panel C).
Next to determine if the transfection of tumour cells with TF-specific siRNA affected their ability to metastasize to bone, we injected two groups of C57BL/6 mice with either TF siRNA-transfected or mock-transfected td-tomato expressing tumour cells. A third group of mice served as an age-matched control and did not receive any tumour cells. Two weeks later, the mice were sacrificed, their femurs removed, and td-tomato fluorescence was used to determine the presence or absence of tumour cells. As seen in Figure 2A and B, fluorescent cells were present in the femurs of mice that received either mock-transfected or TF siRNA-transfected tumour cells. However, fluorescence was significantly reduced when the mice were inoculated with TF-specific siRNA-transfected tumour cells rather than mock-transfected tumour cells (Fig. 2B vs. 2A). In contrast, no fluorescence was observed in the femurs of those mice which did not receive tumour cells (Fig. 2C). When quantified, tumour burden in the femurs of mice receiving mock-transfected tumour cells was 9.18 ± 2.20% while the tumour burden in mice receiving TF siRNA-transfected tumour cells was only 2.20 ± 0.58% (P < 0.001) (Fig. 2D).
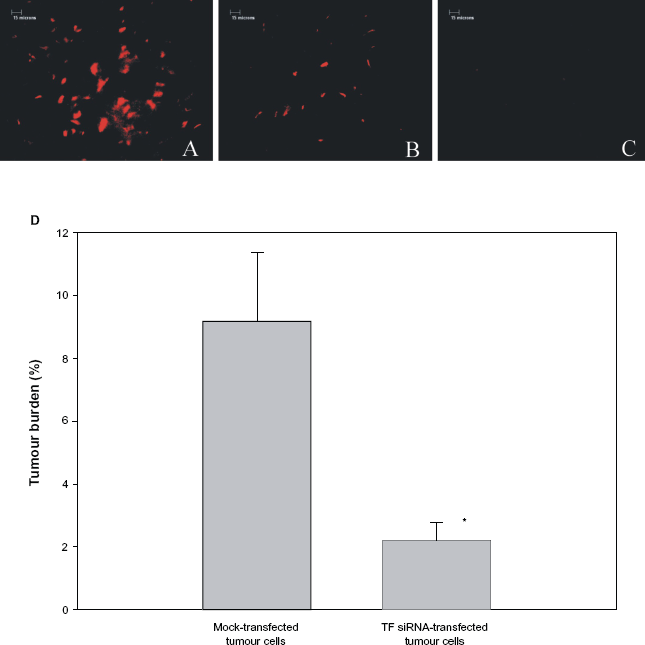
Tumour burden in mice receiving left ventricular injections of TF-specific siRNA-transfected tumour cells. Mice received left ventricular injections of either 5 × 105 mock-transfected B16F10 cells (Panel A), or an equivalent number of TF-specific siRNA-transfected B16F10 melanoma cells (Panel B). One group of mice also served as a control and received no tumour cells (Panel C). On day 14, all animals were sacrificed and tumour burden within the femurs determined by fluorescence using a Mertz grid (Panel D), as described in the Materials and Methods. Bar: 15 microns. Data is expressed as a mean ± SEM.
Cancellous bone volume in mice injected with TF-specific siRNA-transfected tumour cells
In order to determine if the transfection of B16F10 melanoma cells with TF- specific siRNA prevented their ability to cause bone loss, both radiographic analysis and bone histomorphometry were performed. As seen in Figure 3, radiographic analysis of the distal portion of femurs from mice that received mock-transfected tumour cells revealed distinct areas of osteolysis (Fig. 3B). In contrast, no osteolysis was observed in femurs from mice that did not receive tumour cells (Fig. 3A) or mice that received TF specific siRNA-transfected tumour cells (Fig. 3C). In order to quantify these differences in tumour cell-induced bone loss, a region 200 | m below the epiphyseal growth plate that included the entire metaphysis was subjected to bone histomorphometry using a Mertz grid. As shown in Figure 3D, mice that received mock-transfected tumour cells demonstrated a significant reduction in cancellous bone volume (BV/TV) when compared to mice that did not receive tumour cells (12.57 ± 1.59% vs. 25.56 ± 3.22%, respectively; P < 0.005). In contrast, the femurs from mice that received TF siRNA-transfected tumour cells displayed no significant reduction in cancellous bone volume when compared to control mice that did not receive tumour cells (20.51 ± 2.75% vs. 25.56 ± 3.22%; P > 0.05). Thus, cancellous bone loss was prevented when the tumour cells were transfected with TF-specific siRNA prior to being injected into mice.
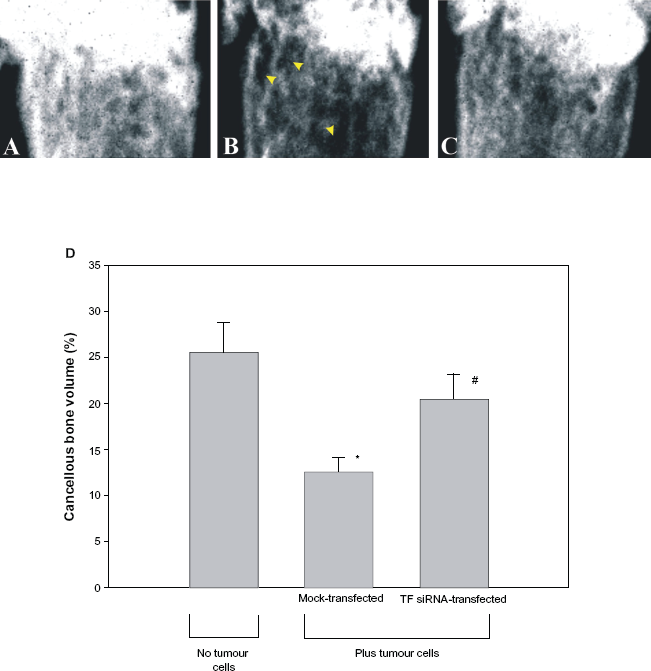
Cancellous bone volume in mice receiving left ventricular injections of TF-specific siRNA-transfected tumour cells. Mice received either no tumour cells (Panel A) or 5 × 105 B16F10 melanoma cells transfected with either mock siRNA (Panel B) or TF-specific siRNA (Panel C). On day 14, the animals were sacrificed and their femurs examined using a Faxitron cabinet X-ray system in order to observe areas of osteolysis (arrows). Alternatively, their left femur was embedded in glycomethacrylate, sectioned, and the distal end examined microscopically using a Mertz grid in order to determine percent cancellous bone volume in each femur (Panel D). Data is expressed as mean ± SEM.
Since a decrease in cancellous bone volume should correspond with a decrease in either trabecular width or number, we next examined how administering TF-specific siRNA-transfected B16F10 murine melanoma cells to mice would affect these parameters as well. As seen in Table 1, mice that were injected with mock-transfected tumour cells demonstrated a significant reduction (P < 0.05) in both trabecular width and trabecular number when compared to mice that did not receive any tumour cells (75.09 ± 7.11 vs. 112.82 ± 7.13 μm; and 7.50 ± 0.96 vs. 11.25 ± 0.85 trabeculae/mm2, respectively). However, no significant difference (P > 0.05) in trabecular width or number was seen when mice that were injected with TF-specific siRNA-transfected tumour cells were compared to mice that did not receive tumour cells (106.72 ± 7.57 vs. 112.82 ± 7.13 μm and 12.50 ± 1.19 vs. 11.25 ± 0.85 trabeculae/mm2, respectively).
Changes in trabecular width, number and separation in mice receiving left ventricular injections of TF-specific siRNA-transfected tumour cells.
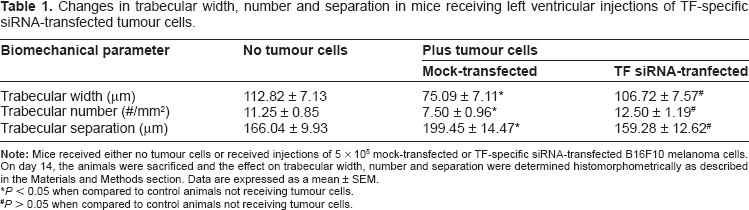
P < 0.05 when compared to control animals not receiving tumour cells.
P > 0.05 when compared to control animals not receiving tumour cells.
Osteoclast surface in mice injected with TF-specific siRNA-transfected tumour cells
To determine if the tumour cell-induced changes in cancellous bone volume were caused by an increase in osteoclast number, osteoclast surface (Oc.S/BS) was quantitated by bone histomorphometry. As shown in Figure 4, the femurs from animals that received mock-transfected tumour cells had a 30.6 ± 7.8% increase in osteoclast surface, as compared to the femurs isolated from mice that did not receive tumour cells (2.22 ± 0.12% vs. 1.54 ± 0.12%, P < 0.005). In contrast, no significant difference (P > 0.05) in osteoclast surface was seen when mice that were injected with TF-specific siRNA-transfected tumour cells were compared to mice that did not receive tumour cells (1.58 ± 0.22% vs. 1.54 ± 0.12%).
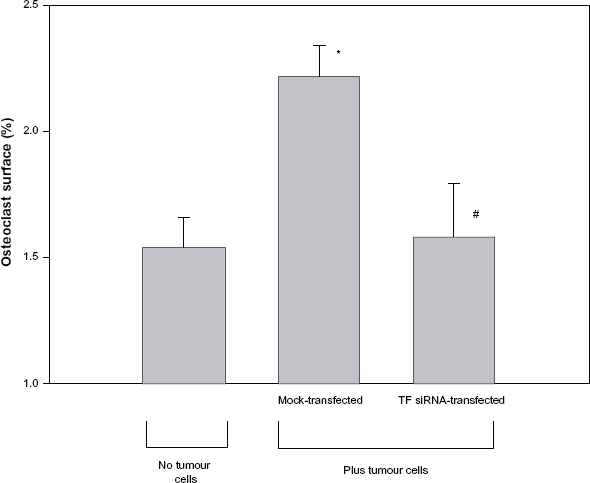
Osteoclast surface in mice receiving left ventricular injections of TF-specific siRNA-transfected tumour cells. Mice received either no tumour cells or received injections of 5 × 105 mock-transfected or TF-specific siRNA-transfected B16F10 melanoma cells. On day 14, all animals were sacrificed and a Mertz grid was used to determine the percentage of cancellous bone surface covered by osteoclasts. Data is expressed as mean ± SEM.
Femoral bone strength in mice injected with TF-specific siRNA-transfected tumour cells
Biomechanical testing was performed in order to determine if the expression of TF by cancer cells affects the biomechanical properties of bone. As shown in Figure 5, the maximum load tolerated by femurs of animals that received mock-transfected tumour cells was 9.3 ± 2.7% lower than the maximum load tolerated by the femurs isolated from animals that did not receive tumour cells (14.59 ± 0.44 N vs. 16.09 ± 0.55 N, P < 0.05). In contrast, no difference in maximum load tolerated was seen when the femurs of mice that received TF-specific siRNA-transfected tumour cells were compared with the femurs of mice that did not receive tumour cells (15.94 ± 0.32 N vs. 16.09 ± 0.55 N, P > 0.05). A similar trend was observed between treatment groups when the biomechanical parameters of stress at maximum load, stiffness, energy to break, toughness and elastic modulus were also compared (Table 2).
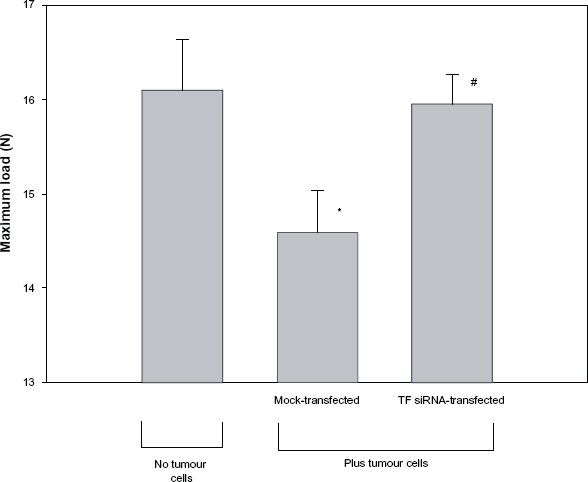
Femoral bone strength in mice receiving left ventricular injections of TF-specific siRNA-transfected tumour cells. Mice received either no tumour cells or received injections of 5 × 105 mock siRNA-transfected or TF-specific siRNA-transfected B16F10 melanoma cells. On day 14, the animals were sacrificed, their femurs removed and subjected to biomechanical testing by three-point bending. Data is expressed as a mean ± SEM.
Changes in the biomechanical parameters of bone following left ventricular injection of TF-specific siRNA-transfected tumour cells.
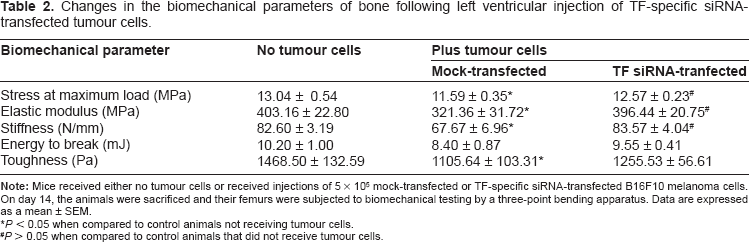
P < 0.05 when compared to control animals not receiving tumour cells.
P > 0.05 when compared to control animals that did not receive tumour cells.
Discussion
In this study, we used a well-defined animal model of osteolytic bone metastasis to show that the ability of B16F10 melanoma cells to metastasize to bone is inhibited when their expression of TF is down-regulated with siRNA treatment. In addition, by performing bone histomorphometry and biomechanical testing, we were able to demonstrate that TF-specific siRNA-transfected tumour cells cause significantly less bone loss than do mock-transfected tumour cells. To our knowledge, this is the first report to show that TF promotes the metastasis of cancer cells to bone.
Numerous studies have shown that B16F10 melanoma cells can cause bone loss.17–19 The current study supports these findings and suggests that B16F10 melanoma cells cause bone loss by increasing the formation of osteoclasts within the bone microenvironment. Thus, we demonstrated significant bone loss and increased osteoclast surface (OC.s/TV) when mice were injected with mock-transfected B16F10 melanoma cells (Figs. 3 and 4). However, when the mice were injected with tumour cells that had been transfected with TF-specific siRNA, fewer cancer cells were found localized within the bone microenvironment and as a result, osteoclast surface was not increased (Figs. 2 and 4). Consequently, mice receiving TF siRNA-transfected tumour cells did not demonstrate a loss in cancellous bone volume (Fig. 3) or a decrease in femoral bone strength (Fig. 5). When taken together, these findings suggest that the cancer cells themselves are responsible for both the bone loss and the increase in osteoclast number that we observed when mice were injected with mock-tranfected tumour cells.
Our results suggest that TF expression by murine melanoma cells plays an important role in their ability to metastasize to bone. Femurs from mice receiving tumour cells with reduced TF expression (TF siRNA-transfected tumour cells) display reduced tumour burden when compared to those from mice that received tumour cells with normal TF expression. How TF acts to promote the metastasis of cancer cells to bone is unknown. However, several mechanisms have been proposed to account for TF's ability to promote pulmonary metastasis. For example, TF expression on the surface of tumour cells has been shown to result in thrombin generation. This, in turn, has been proposed to cause endothelial cell activation, as well as platelet aggregation and fibrin deposition.20,21 Thus, it is possible that TF expression by tumour cells promotes their ability to metastasize to bone by activating bone-derived endothelium to express tumour cell- specific adhesive moieties such as P-selectin.15,16 Alternatively, thrombin generated from the TF pathway may also lead to local fibrin generation and platelet activation on the cancer cell surface, which could serve to protect the cancer cells from host defense mechanisms. In support of this latter possibility, Palumbo et al demonstrated that circulating tumour cells were protected from natural killer (NK) cells when they formed emboli with platelets and fibrin.22,23
Aside from its ability to cause thrombin generation, TF itself can act to induce signal transduction when bound to FVIIa. Numerous studies have shown that TF/FVIIa complexes can initiate signal transduction by binding to a family of surface receptors called protease-activated receptors or PARS. PARS are G-protein-coupled receptors that are activated by proteolytic cleavage and are expressed on several cell types. TF/FVIIa complexes are able to activate PAR-2, whereas TF/FVIIa/FXa complexes can activate both PAR-1 and PAR-2. 24 Both PAR-1 and PAR-2 receptors are expressed on tumour cells. 25 Thus, the intracellular signalling initiated by the TF/FVIIa complex may influence several early prometastatic events, such as tumour cell adhesion or migration.26–28 In addition, several studies have suggested that the intracellular signaling initiated by TF/FVIIa complexes can promote metastasis by inducing tumour cell survival. In support of this hypothesis, two independent studies have shown that FVIIa promotes the survival of BHK cells in the absence of serum if the cells are first transfected with TF.29,30 Both studies observed that FVIIa's protective function was dependent on its proteolytic activity, and that this anti-apoptotic effect appeared to involve TF/FVIIa's ability to suppress caspase-3 activation as well as activate the anti-apoptotic kinases, p44/42 MAP kinase and PI3 kinase.29,30
While TF has been shown to increase tumour cell survival, previous studies have failed to show any effect of TF on tumour cell proliferation in vitro.31,32 In agreement with these findings, studies performed in our laboratory failed to demonstrate any differences in cellular proliferation when untransfected or mock-transfected B16F10 melanoma cells were compared to TF-specific siRNA-transfected tumour cells over time (data not shown). Regardless of the exact mechanism by which TF elicits its prometastatic effects, our results suggest that TF is initiating its effects on tumour cell metastasis early on in the process. Thus, while we were able to knockdown TF mRNA expression in murine melanoma cells by approximately 80%, this effect was transient with TF expression returning to normal within 5 days following transfection (Fig. 1B). Similar findings were reported by Amarzguioui et al. 11
In the current study, transiently transfected cells were used instead of stably transfected cells because the selection of a stably transfected cell line would necessitate the clonal expansion of the transfected cell line from a single cell. Tumour cells are typically heterogeneous in their properties. Therefore if we had stably transfected our cells with TF-specific siRNA, we would not have been able to determine if the reduction in metastatic ability was due to the stable knockdown of TF or if our population of transfected cells had arisen from a single cell that just happened to have low metastatic ability. Therefore, we thought it would be best to use transiently transfected cells so that the only difference between cell populations was their expression of TF.
The results of this study may help to explain our earlier findings that heparin inhibits the ability of cancer cells to metastasize to bone. 17 Heparin is a commonly used anticoagulant that acts primarily to inhibit thrombin by promoting its interaction with a naturally occurring inhibitor called antithrombin. Heparin is also known to induce tissue factor pathway inhibitor (TFPI) release from endothelial cells. TFPI is known to inhibit TF activity by binding to TF/FVIIa complexes in a FXa-dependent manner. 8 Given that the results of the current study demonstrate that TF promotes the ability of cancer cells to metastasize to bone, it is possible that heparin's anti-metastatic properties may reflect its ability to inhibit the TF pathway. This would help explain why median survival times and survival rates are significantly higher in those patients who receive heparin and chemotherapy as opposed to those who receive chemotherapy alone. 33
In summary, we demonstrate that the ability of B16F10 melanoma cells to metastasize to bone is inhibited when TF expression is down-regulated in cancer cells by siRNA treatment. In addition, by performing bone histomorphometry and biomechanical testing, we were able to demonstrate that TF-specific siRNA-transfected tumour cells cause significantly less bone loss than do mock-transfected tumour cells. This suggests that TF-specific siRNA, or other small molecule inhibitors of TF, may have the potential to be used clinically as a therapeutic agent for preventing cancer metastasis to bone.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgement of Funding Sources
Supported by a Canadian Institutes of Health Team Grant in Venous Thromboembolism (#MOP-FRN-79846). A.P is a holder of an Ontario Graduate Scholarship (OGS).