
Abstract
The extracellular matrix glycosaminoglycan hyaluronan plays a key role in the development and pathogenesis of malignant disease. Reflecting its functional importance, the molecule is expressed at greatly elevated levels within many solid tumors. Although little explored, differences in the level of hyaluronan present in normal and malignant tissues could potentially be exploited to more effectively target gene therapy to tumor sites in vivo. As a first step toward this goal, we describe here a family of chimeric proteins in which the extracellular ligand-binding domain of the hyaluronan receptor CD44 is fused in-frame to the cytoplasmic “death domain” of the pro-apoptotic protein Fas. Although these chimeric proteins can be stably expressed on the surface of transduced tumor cells in the absence of hyaluronan, upon interaction with the ligand, apoptosis is rapidly induced. Both exogenous and endogenous tumor produced hyaluronan can function as triggers, dramatically reducing clonogenic potential. Together, these studies help validate a broadly applicable gene therapy approach in which the presence of particular multivalent ligands within the tumor microenvironment can be exploited for therapeutic gain.
Introduction
It has long been appreciated that the glycosaminoglycan hyaluronan (HA) is selectively enriched within solid tumors, most notably in peritumoral regions.1–3 The potential importance of this finding with respect to the development and/or pathogenesis of malignant disease is emphasized by the key role played by HA in the regulation of various cell migration, proliferation and differentiation events, particularly during embryonic development. 4 Specifically, HA is highly hydroscopic and can adopt a pseudo-random coil-like configuration that occupies large volumes in solution such that, when present at high concentrations, the molecule can disrupt the organized architecture of tissues, opening spaces through which cells can migrate more easily.5,6 Conversely, a reduction in HA expression often correlates with the cessation of cell movement and the induction of differentiation. In adults, temporal alterations in the level of HA are seen in the course of tissue injury and repair, 7 while in the case of malignant disease, correlations have been established between HA levels and both local invasion and metastasis. 8 An association between HA levels and disease recurrence and prognosis has also been described in a number of studies.1,3,9,10 While the mechanisms by which HA mediates these diverse effects on tumor growth remain to be fully defined, it is evident that the molecule functions in both a direct and indirect fashion, altering tumor cell proliferation and differentiation while also enhancing the recruitment of stromal cells and stimulating angiogenesis.6,11,12
The biological effects triggered by the presence of HA are mediated by a number of cell surface and soluble HA-binding proteins.13–19 Among these, members of the CD44 family appear to be particularly important with regard to the development and pathogenesis of malignant disease.20–23 The CD44 isoform present on most resting and non-transformed cells, designated CD44H or CD44s, has a molecular weight in the range of 80–90 kD. However, alternative splicing events involving a contiguous series of 10 or more exons (v1–v10) present within the single copy CD44 gene can generate a large number of higher molecular weight isoforms that are differentially expressed and may be present at elevated levels on certain activated and/or malignant cell types. 24 Site-directed mutagenesis studies have suggested that a motif containing two basic amino acids separated by a stretch of 7 non-acidic amino acids (B[X7]B) facilitates binding to HA.25,26 This motif is found in all HA-binding proteins that have been examined to date and occurs twice within the extracellular domain of CD44 in a region shared by all alternatively spliced isoforms.25,27 An additional B[X7]B motif is also present within the sequence encoded by the alternatively spliced exon v10 and plays a key role in mediating adhesive interactions between v10 containing CD44 isoforms. 27
The upregulation of HA within malignant tissues raises the possibility that differential expression of the molecule could perhaps be exploited in the development of tumor targeted therapy. The approach we have taken in this regard is to generate a family of chimeric proteins in which the extracellular ligand binding domain of the HA receptor CD44 is fused in-frame to the intracellular “death-domain” of the pro-apoptotic protein Fas (CD44/Fas). It was hypothesized that rather than triggering the signal transduction events normally induced upon the binding of HA by CD44, when expressed in target cells, interaction of the chimeric CD44/Fas protein with HA would instead induce apoptotic death.
The results obtained confirm the potential utility of this approach. Specifically, chimeric CD44/Fas proteins proved effective at binding HA and in triggering apoptosis in transduced target cells upon interaction with ligand. It is proposed that chimeric proteins of this type may prove useful in the context of cancer gene therapy.
Materials and Methods
Cell lines
The tumor cell lines K562 (erythroleukemia), T24 (transitional bladder carcinoma), MCF7 (mammary adenocarcinoma) and PC3 (metastatic prostate adenocarcinoma) were obtained from the American Type Culture Collection (Manassas, VA). 293 cells (adenovirus 5 transformed human embryonic kidney cells) were obtained from Qbiogene (Carlsbad, CA). All cell lines were incubated at 37 °C in an atmosphere containing 5% CO2 in Dulbecco's Minimal Essential Medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin sulfate (DMEM + 10% FBS).
Monoclonal antibodies
Two mouse monoclonal antibodies (mAbs) directed against human Fas were used in the course of this study. The first, designated DX2 (BD Biosciences, San Jose, CA), is directed against an epitope present on the extracellular domain of Fas and thus recognizes endogenous Fas but not the CD44/Fas chimeras described below that contain only the transmembrane and/or cytoplasmic domain of the molecule. The second, mAb 3D5 (Alexis Biochemicals, San Diego, CA), defines an epitope encoded within the death domain of Fas and thus recognizes both endogenous Fas and the CD44/Fas chimeras.
The generation and characterization of the anti-CD44 mAb 4A4 has been described in detail elsewhere.28,29 The phycoerythrin-conjugated anti-CD44 mAb G44-26 (CD44-PE) was purchased from BD Biosciences.
Expression vectors
The EBV-based episomal expression vector pCEP4 was purchased from BD Biosciences. The major features of both pCEP4 and the cDNA constructs used in this study are shown in Figure 1.
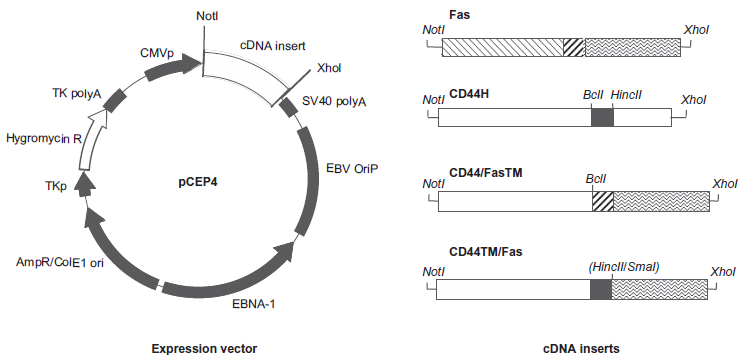
Major features of the pCEP4 vector and cDNA constructs used in this study. The two CD44/Fas chimeras differ from one another only with respect to the origin of their membrane spanning domains. Thus the membrane spanning domain present in CD44/FasTM is derived from Fas, while that present in CD44TM/Fas is derived from CD44H.
Two chimeric proteins designated CD44/FasTM and CD44TM/Fas were generated for use in this study. As shown in Figure 1, both contain the extracellular HA-binding domain of CD44 and the cytoplasmic domain of Fas that includes the so-called “death domain” that triggers an apoptotic response upon receptor ligation. Where the two constructs differ is with respect to the sequences used to join these two functionally important regions. Thus, CD44/FasTM incorporates the membrane spanning domain of Fas, while in CD44TM/Fas the membrane spanning domain of CD44 is instead used (Fig. 1).
To generate the vector pCEP4.CD44H, a fragment containing a full-length cDNA encoding the 90 kD “hemopoietic” or “standard” isoform of CD44 (CD44H/CD44s) was released from the plasmid pCDM8.CD44 clone #2.730 by digestion with HindIII and Notl and ligated into the HindIII-Notl sites of pCEP4.
To construct pCEP4.Fas, a full length human Fas cDNA (~1.5 kb) was first generated by PCR using Jurkat cDNA as a template and the following primer pair: 5′ Fas UTR (5′-GCGGAATTCAGGGGCGGGCACTGGCAC-3′) and 3′ Fas UTR (5′-GGCTCGAGAATCTTTTCAAACACTAATTGC-3′) containing respectively engineered 5′ EcoRl and Xhol sites. After 40 cycles (95 °C for 30s, 60 °C for 30s, 72 °C for 2.5 min), the resultant PCR products were separated by gel electrophoresis and a species of the appropriate size isolated, digested with EcoRl and Xhol and cloned into EcoRl-Xhol digested pBluescript II (KS+) generating vector pBS.Fas. The insert was subsequently sequenced and proved to be identical to the published Fas sequence with the exception of an A to G substitution at position 1117 resulting in a single amino acid change at residue 297, that converts a Glu residue within the cytoplasmic domain of the mature protein to Gly. Finally, the full-length Fas cDNA was isolated from pBS.Fas by digestion with Notl and Xhol and the resultant fragment cloned into the Notl-Xhol sites of pCEP4.
pCEP4 vectors encoding the CD44/Fas chimeras CD44/FasTM and CD44TM/Fas (Fig. 1) were generated as follows:
Construction of the CD44TM/Fas chimera
In order to acquire the restriction sites needed to facilitate subsequent cloning steps, a fragment containing the full-length CD44H cDNA was first released from the pCEP4.CD44H vector described above by digestion with Xhol. The ends were blunted/filled-in using T4 DNA polymerase and the fragment cloned into the EcoRV site of plasmid pZErO2 (Invitrogen, Carlsbad, CA). After checking for orientation, the full-length CD44H cDNA was once again isolated by digestion with Notl-EcoRl and cloned into Notl-EcoRl digested pBluescript II (KS+) generating plasmid pBS.CD44H. Finally, the full-length CD44H cDNA was again released, this time by digestion with Notl and Xhol, gel purified, then partially digested with HincII generating a Notl-HincII (blunt) fragment that contains the extracellular and membrane spanning domains of CD44H (CD44TM).
A cDNA encoding the cytoplasmic domain of human Fas was generated by PCR using the pCEP4.Fas plasmid described above as a template and the following primer pair: 5′-Fas CYTO2 (5′-GCCCGGGGTGAAGAGAAAGGAAGTAGAG-3′) and 3′-Fas UTR (5′-GGCTCGAGAATCTTTTCAAACACTAATTGC-3′) containing respectively engineered 5′ Smal and Xhol sites. After 35 cycles (95 °C for 30s, 60 °C for 30s, 72 for 1 min), the resultant PCR products were separated by gel electrophoresis and a fragment of the appropriate size isolated. The ends were repaired/filled-in with T4 DNA polymerase and the fragment cloned into the EcoRV site of pZErO2. After checking for appropriate (i.e. reverse) orientation, the resultant vector was sequentially digested with Notl then with Smal and the linearized plasmid ligated with the Notl-HincII fragment containing the extracellular and membrane spanning domains of CD44H generated as described above, producing the fulllength CD44TM/Fas chimera with a Notl site at its 5′ end and a Xhol site at its 3′ end. For expression studies, the chimeric cDNA was isolated from the pZErO2 vector by digestion with Notl and Xhol and cloned into the Notl-Xhol sites of pCEP4.
Construction of the CD44/FasTM chimera
A cDNA encoding the membrane spanning and cytoplasmic domains of human Fas was generated by PCR using pCEP4.Fas plasmid DNA as a template and the following primer pair: 5′-Fas TM (5′-AACGTGAT- CATCCTTTGTCTTCTTCTTTTG-3′) and 3′-Fas UTR (5′-GGCTCGAGAATCTTTTCAAACACTAATTGC-3′) containing respectively engineered 5′ Bcll and Xhol sites. After 35 cycles (95 °C for 30s, 60 °C for 30s, 72 °C for 1 min), the resultant PCR products were separated by gel electrophoresis and a fragment of the appropriate size isolated. The ends were repaired/filled-in with T4 DNA polymerase and the fragment cloned into the EcoRV site of pZErO2. After identifying a clone in which the insert is present in the appropriate (i.e. reverse) orientation, a fragment containing the membrane spanning and cytoplasmic domain of Fas (FasTM) was isolated by digestion with Bcll-Xhol. The pBS.CD44H vector generated as described above was also digested with Bcll and Xhol thereby removing sequences encoding the membrane spanning and cytoplasmic domains of CD44H. The resultant vector was ligated with the Bcll-Xhol FasTM fragment producing the full-length CD44/FasTM chimera with a Notl site at its 5′ end and a Xhol site at its 3′ end. The first 5 amino acids in the transmembrane domain of the CD44/FasTM chimera correspond to CD44H while the remainder of the membrane spanning domain and the entire cytoplasmic domain correspond to Fas. Finally, the chimeric cDNA was isolated from the pBS vector by digestion with Notl and Xhol and cloned into the Notl-Xhol sites of pCEP4.
Transfection of K562 cells
An electroporation technique was used to introduce pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4.CD44/ FasTM or pCEP4.CD44TM/Fas plasmid DNA into K562 cells. Briefly, log-phase K562 cells were harvested, washed several times in phosphate buffered saline (PBS) before being resuspended in PBS at 1–2 × 107 cells/ml. Twenty μg of each plasmid DNA were added to 4 × 106 cells and the cell suspensions transferred to 2 mm gap cuvettes and electroporated using the BTX ECM 600 Electroporator System (BTX, San Diego, CA). The settings were 280 V, capacitance 500 μF, and resistance 48 Ω. The time constants obtained generally ranged from 3.3 to 3.5 msec. Immediately after electroporation, the cells were diluted with 35 ml DMEM + 10% FBS and plated in 15 cm Integrid tissue culture dishes (Falcon). Hygromycin B (Sigma) was added 24 h later at a final concentration of 250 μg/ml to select for cells that had taken up and expressed plasmid DNA.
FACS analysis
K562 cells transfected with pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4.CD44/FasTM or pCEP4. CD44TM/Fas plasmid DNA and selected and maintained in hygromycin (250 μg/ml) were examined for CD44 and Fas expression by FACS analysis. Briefly, approximately 2 × 105 cells were incubated on ice for 30 min with either the anti-Fas mAb DX2 or the anti-CD44 mAb 4A4 at a final concentration of approximately 2 μg/ml. Following extensive washing in Hank's balanced salt solution (HBSS) containing 2% FBS (HBSS + 2% FBS), the cells were incubated for a further 30 min on ice with FITC-conjugated rabbit anti-mouse Ig (2 μg/ml) (BD Biosciences). After additional washing, the cells were resuspended in HBSS + 2% FBS containing 1 μg/ml propridium iodide to facilitate the identification and exclusion of dead cells and analyzed on a FACSCalibur (BD Biosciences, Immunocytometry Systems, San Jose, CA).
HA binding assay
The HA-binding activity of K562 cells transfected with pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas was determined by FACS analysis. For each line, approximately 2 × 105 cells were resuspended in 0.5 ml of HBSS + 2% FBS), a 1:100 dilution of anti-CD44 mAb PEG44-26 (BD Pharmingen), and a 1:1000 dilution of fluorescein isothiocyanate-conjugated HA (FITC-HA, ~50 μg/ml final), and the mixture was incubated for 1 h on ice. Following repeated washing with HBSS + 2% FBS, cells were resuspended in HBSS + 2% FBS containing 1 μg/ml propridium iodide and analyzed on a FACSCalibur (BD Biosciences Immunocytometry Systems).
Induction and measurement of apoptosis
Ten cm non-tissue culture Petri dishes (Falcon) were coated with HA by adding 5 ml of a solution of potassium hyaluronate purified from human umbilical cord (Sigma) (5 mg/ml in PBS) and incubating overnight at 4 °C. Unbound HA was removed by extensive washing and dishes carefully drained. Approximately, 1 × 107 K562 cells transfected with either pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4. CD44/FasTM or pCEP4.CD44TM/ Fas in a final volume of 3 ml HBSS were added to each HA-coated plate and the cells allowed to settle and interact with the immobilized HA for approximately 30 min. All of the cells, whether adherent or not, were then harvested by gentle pipetting, washed 3 times with PBS, resuspended in 50 ml DMEM + 10% FBS and the cell suspension added to a 125 cm2 tissue culture flask (Falcon). Flasks incubated at 37 °C and at various time points aliquots of cells were removed, washed with PBS and the cell pellets gently resuspended in 5 ml 70% ethanol. Samples were stored at –20 °C until all time points had been collected. The cells were then spun down and the percentage of hypodiploid apoptotic cells determined after staining in a PBS solution containing propridium iodide and RNase A.
Western blot analysis
293 cells were electroporated with pCEP4, pCEP4. Fas, pCEP4.CD44H, pCEP4.CD44/FasTMorpCEP4. CD44TM/Fas plasmid DNA, harvested 2 days later by incubation with PBS containing 3 mM EDTA, and the expression of both CD44 and Fas determinants examined by Western blot. Briefly, cells were washed in PBS and their membranes solubilized by incubating the pellets for 15 min on ice in 10 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA, l% NP-40 at ~2 × 107 cells/ml lysis buffer. Nuclei and detergent insoluble material were removed by centrifugation and the lysates stored at –80 °C until required. Thawed samples were added to an equal volume of non-reducing sample buffer (125 mM Tris, 20% (v/v) glycerol, 4.6% (w/v) SDS, pH 6.8) and incubated at 100 °C for 5 min. Total cellular proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes. The filters were incubated overnight at 4 °C in PBS containing 5% (w/v) milk protein then probed with either mAb 4A4, which recognizes an epitope present in the extracellular domain of CD44, or mAb 3D5, directed against the death domain of Fas (2 μg/ml in HBSS + 2% FBS). After extensive washing, filters were incubated for a further 1–2 hr with a 1:100 dilution of horse radish peroxidase (HRP)-conjugated rabbit anti-mouse Ig secondary antibody. After additional washing, the filters were developed in ECL solution (Amersham Pharmacia Biotech) and protein products imaged on photographic film (Amersham Life Sciences, Buckinghamshire, UK).
Clonogenic survival assay
The effect of CD44/FasTM and CD44TM/Fas expression on the long-term survival and proliferation of tumor cell lines was determined using a standard clonogenic assay. pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas plasmid DNA was introduced into 293, T24, PC3 and MCF7 cells by electroporation as follows. Tumor cells were harvested by trysinization and resuspended in PBS at 1–2 × 107 cells/ml. Twenty μg of plasmid DNA were added to 4 × 106 cells and the cell suspensions transferred to 2 mm gap cuvettes and electroporated using the BTX ECM 600 Electroporator System (BTX, San Diego, CA). For 293 and MCF7 cells a voltage of 270 V, capacitance of 400 μF and resistance setting of 72 Ω were used. For T24 and PC3 cells 280 V and a resistance of 48 Ω were employed with capacitance settings of 400 μF and 300 μF respectively. Time constants generally ranged from around 3.0 to 3.5 msec for 293, MCF7 and T24, and 2.0–2.2 msec for PC3. Immediately after electroporation, the transfected cells were transferred into 35 ml DMEM + 10% FBS and plated in 15 cm Integrid tissue culture dishes (Falcon). Hygromycin B (Sigma) was added 24 h later to a final concentration of either 200 μg/ml (293, PC3 and MCF7) or 250 μg/ml (T24) to select for cells that had taken up and expressed plasmid DNA. Around 10–14 days later, plates were fixed and stained with a methylene blue/methanol solution and the number of hygromycin-resistant colonies determined.
Results
For the chimeras described in this study to be of use in cancer therapy, it is important that the encoded proteins are stable and can be expressed on the surface of transduced cells at a level sufficient to produce a biological effect upon binding of ligand. In order to demonstrate that this was indeed the case, both the CD44/FasTM and CD44TM/Fas cDNA constructs were cloned into the EBV-based episomal plasmid vector pCEP4 (Fig. 1) and introduced into CD44-negative K562 cells by electroporation. Transfectants were selected in the presence of hygromycin and surface expression of the corresponding chimeric proteins determined by FACS analysis using an antibody directed against the extracellular domain of CD44. Full length CD44H and Fas cDNAs were used as controls. As shown in Figure 2, in the absence of added ligand, K562 cells expressing high levels of CD44/FasTM or CD44TM/Fas could be readily established and maintained for extended periods of time in vitro without apparent loss of viability. Interestingly, although the two Fas chimeras were expressed at a similar level on the surface of transduced K562 cells (as determined by mean fluorescence intensity), the level of wild-type CD44 was generally higher, even though the same vector system and transfection procedures were used.
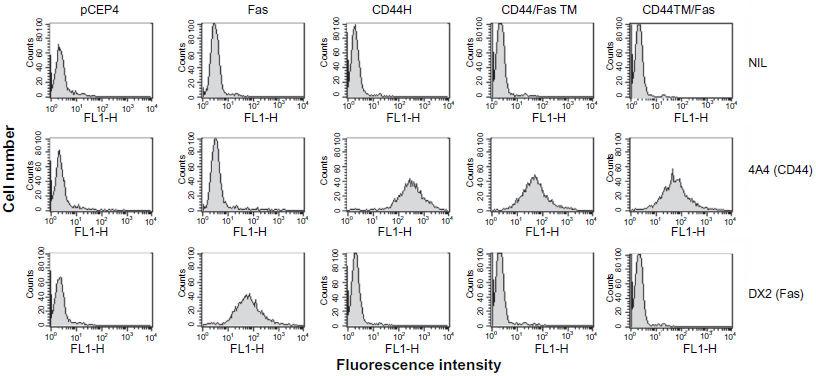
Expression of chimeric CD44/Fas proteins on the surface of transduced tumor cells. pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas plasmid DNAs were introduced into CD44-negative K562 tumor cells by electroporation. pCEP4, pCEP4.Fas and pCEP4.CD44H plasmids were used as controls. Transfected cells were selected in the presence of Hygromycin B and the expression of CD44 and Fas on the surface of established lines determined by FACS analysis after staining respectively with mAb 4A4 or mAb DX2.
To ensure that the chimeric proteins are processed in an appropriate manner, pCEP4 vectors encoding the CD44/FasTM and CD44TM/Fas cDNAs were introduced into 293 cells by electroporation and 24 hr later the transfected cells were harvested, membrane lysates prepared and Western blots run and probed with mAbs directed against either CD44 (mAb 4A4) or the the death domain of Fas (mAb 3D5). 293 cells were specifically chosen for these studies as they are not only CD44-positive but express primarily CD44H (MW 80–90 kD) and not higher molecular weight isoforms, allowing the proteins encoded by the two transgenes to be compared in size and relative expression to endogenous CD44H. As shown in Figure 3, the results obtained confirm the presence within transfected cells of proteins with an apparent molecular weight of around 80–90 kD that react with both the anti-CD44 and anti-Fas death domain antibodies.
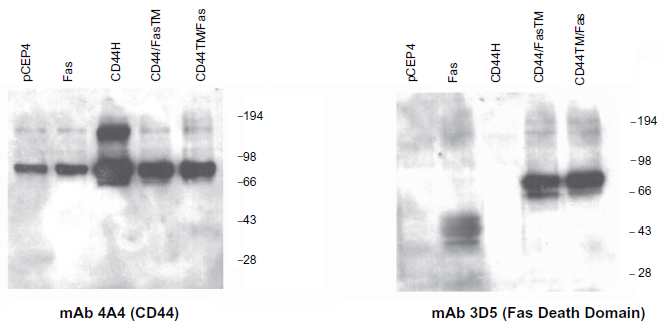
Western blot analysis of CD44/Fas expression in transduced tumor cells. 293 cells were electroporated with pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas plasmid DNA, harvested 2 days later, and the expression of both CD44 determinants and Fas determined by Western blot analysis. Lysates were run on a 7.5% SDS-PAGE gel (2 × 105 cell equivalents per lane), transferred to nitrocellulose, and probed with mAbs directed against the extracellular domain of CD44 (mAb 4A4) or the death domain of Fas (mAb 3D5). After extensive washing, filters were incubated with and appropriate HRP-conjugated secondary antibody, developed in an ECL solution and protein products imaged on photographic film.
Low levels of a higher molecular weight species of around 160–180 kD corresponding to CD44 homodimers are evident on blots probed with the CD44 mAb 4A4 (Fig. 3). Expression of this species is dramatically upregulated in cells transfected with CD44H but remains low in cells transfected with CD44/FasTM or CD44TM/Fas (Fig. 3).
Monomeric Fas itself has a molecular weight of between 37–50 kD depending on the nature and extent of post-translational modification. While a diffuse species of this size is present in cells transfected with pCEP4.Fas, little if any expression is evident in control 293 cells or in 293 cells transfected with pCEP4. CD44H, pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas (Fig. 3).
The CD44/Fas chimeras were next tested for their HA-binding ability. Transfected K562 cells expressing wild type CD44H or either CD44/FasTM or CD44TM/Fas were double-labeled with anti-CD44-PE and FITC-HA allowing the relationship between CD44 levels and the amount of HA bound to be determined. The results obtained indicate that cells need to express a certain critical threshold level of CD44 before HA binding can occur. Above this threshold there is linear relationship between CD44 expression and the amount of HA bound (Fig. 4). Importantly, although the threshold level needed to permit HA binding may be slightly lower for CD44H than for either CD44/FasTM or CD44TM/Fas, both chimeras are able to bind HA when expressed at modest levels (Fig. 4).
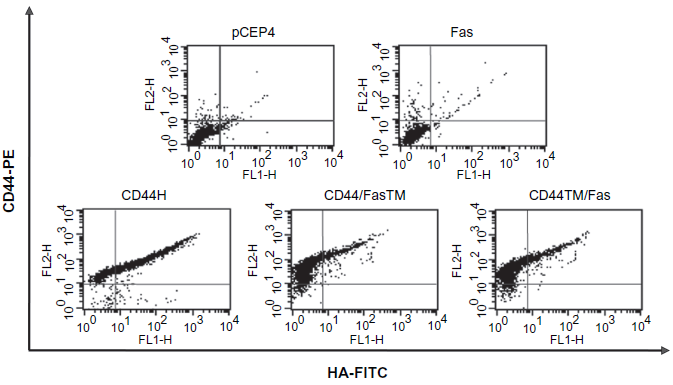
HA-binding activity of chimeric CD44/Fas proteins. K562 cells transfected with pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas were incubated for 1 hr on ice in a solution of HBSS + 2% FBS containing a 1:100 dilution of the anti-CD44 mAb PE-G44-26 (BD Pharmingen) and a 1:1000 dilution of FITC-HA (~50 μg/ml final). Following repeated washing with HBSS + 2% FBS, cells were resuspended in HBSS + 2% FBS containing 1 μg/ml propridium iodide and analyzed on a FACSCalibur (BD Biosciences, Immunocytometry Systems, San Jose, CA).
To determine whether the CD44/Fas chimeras can trigger an apoptotic response upon binding of ligand, K562 cells expressing CD44H or either CD44/FasTM or CD44TM/Fas were allowed to adhere to plastic surfaces coated with HA and the percentage apoptotic (i.e. hypodiploid) cells determined at various time points thereafter. As shown in Figure 5, although CD44H expressing cells bind avidly to HA, they remain largely viable during a 12 hour period of observation. In contrast, a large proportion of K562 cells expressing either of the two CD44/Fas chimeras undergo apoptosis upon binding to HA. Although the ultimate magnitude of the response is similar for both CD44/FasTM and CD44TM/Fas, the rate of apoptosis was consistently greater for cells expressing CD44/FasTM, with near maximal cell death evident as early as 1.5 hours after binding to HA (Fig. 5). By 3 hours, however, the percentage of apoptotic cells was more or less equivalent for both CD44/FasTM- and CD44TM/Fas.
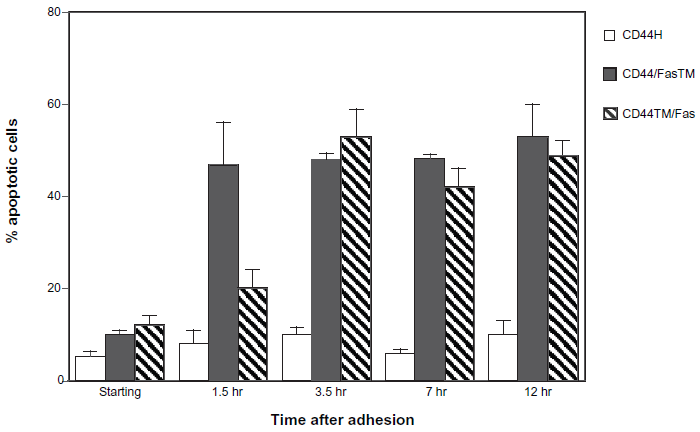
Induction of apoptosis upon interaction of CD44/Fas chimeras with HA. K562 cells transfected with either pCEP4, pCEP4.Fas, pCEP4. CD44H, pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas were added to HA-coated dishes and allowed to settle and interact with the immobilized ligand for approximately 30 min. Both adherent and non-adherent cells were then harvested, resuspended in DMEM + 10% FBS and cultured. At various time points aliquots were removed and the percentage of hypodiploid apoptotic cells determined by FACS analysis after fixation in 70% ethanol and staining in a solution of propridium iodide and RNase A. The data shown represents the mean ± SEM of triplicate cultures.
Finally, the CD44/Fas chimeras were tested for their ability to kill various epithelial tumor cell types that constitutively produce HA. As shown in Figure 6, both CD44/Fas chimeras produced dramatic reductions in the clonogenic potential of the tumor cell lines tested when introduced and expressed by electroporation. There was no significant difference in their relative efficacy as determined using this particular assay system. In agreement with previous studies, wild-type Fas was also effective at killing tumor cells that constitutively express Fas-ligand (FasL) (e.g. 293, MCF-7 and PC-3) but had no effect on the survival and proliferation of the FasL-negative cell line T24 (Fig. 6). Interesting, overexpression of CD44H, although far less effective than the corresponding CD44/Fas chimeras, had a modest inhibitory effect on the clonogenic potential of all four of the tumor lines.
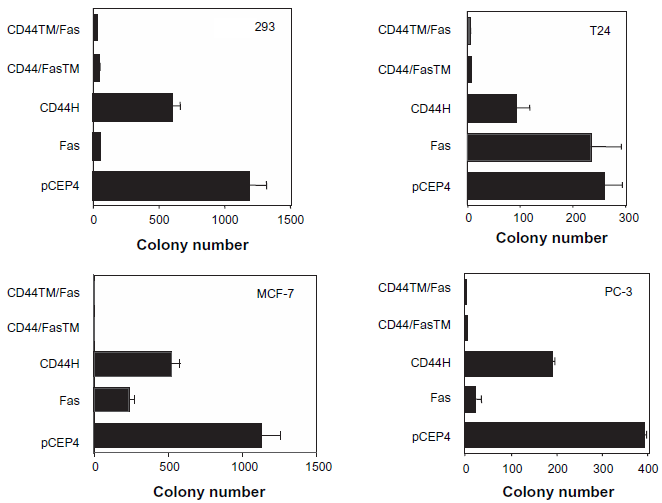
Effect of CD44/Fas expression on the clonogenic survival of tumor cells. 293, MCF-7, PC3 and T24 tumor cells were electroporated with either pCEP4, pCEP4.Fas, pCEP4.CD44H, pCEP4.CD44/FasTM or pCEP4.CD44TM/Fas plasmid DNA and plated in 15 cm Integrid tissue culture dishes. Cells that had taken up plasmid DNA were selected in the presence of Hygromycin B and the ability of transfectants to survive and generate colonies determined on day 10–14 after plates were fixed and stained in a solution of methanol/methylene blue. The colony count data shown represents the mean ± SEM of triplicate cultures.
Discussion
Selective targeting of tumor tissues is key to the success of cancer gene therapy, resulting in increased efficacy while minimizing toxicity. While considerable progress has been made with respect to gene targeting through the use of viral or other vectors with restricted tropism and/or promoter or other regulatory elements that can direct gene expression to desired cell types, there remains considerable room for improvement. Efficient in vivo targeting in particular has proven problematic. Recently we have advocated a novel functional targeting strategy in which the differential presence of various cytokines or extracellular matrix proteins within the tumor microenvironment can be exploited to trigger a therapeutic effect such as cytotoxicity or immune stimulation. The basic approach involves the use of chimeric proteins which, in their simplest form, incorporate both a receptor domain that can recognize and bind a differentially expressed ligand and an effector domain that can translate such binding into a biological effect. In the present manuscript we describe one such family of chimeric proteins in which the extracellular domain of the adhesion protein CD44 is fused in frame to the death domain of the pro-apoptotic protein Fas. These molecules were designed to induce the death of transduced cells upon interaction with the CD44 ligand HA.
Although by no means restricted solely to malignant tissues, HA levels are often greatly elevated within solid tumors.23,31–33 This association is in agreement with a large body of data suggesting an important role for HA in regulating the proliferation, differentiation and migration of both normal and malignant cells.5,34 Increasing HA production by over expression of the HA synthase enzyme, HAS2, enhanced the growth of fibrosarcoma and breast cancer xenografts in nu/nu mice.35,36 Similar results were obtained when the HA synthase HAS3 was overexpressed in human prostate tumor cells, 37 while transfection of antisense Has2 and Has3 reduced the growth and vascularization of prostate tumors in vivo. 38 In keeping with this finding, studies have shown that metastatic colon carcinoma cells express higher levels of HAS3 and produce more HA than equivalent cells isolated from a primary tumor. 39 Later work from the same group confirmed that pericellular HA is necessary for colon carcinoma cell invasion and that this process is critically dependent upon the interaction of HA with CD44. 40
In addition to direct effects on tumor growth and metastasis, signals transduced upon the interaction of CD44 with HA also enhance the resistance of tumor cells to various chemotherapeutic agents including doxorubicin, taxol, vincristine, and methotrexate. 23
This potentially important mechanism may help explain the differential ability of CD44-positive cancer stem cells to survive drug treatment. 23 In support of this conclusion, a number of studies have shown that exposure to hyaluronidase can improve the efficacy of cancer drug treatment.41–43 Blocking the HA-binding activity of CD44 is also effective.23,44,45 Although the mechanisms responsible for this effect remain to be fully elucidated, it has been reported that the interaction between CD44 and HA can regulate the expression and/or function of a number of transporter proteins that mediate drug efflux including MRP2,46,47 P-glycoprotein 46 and BCRP. 48 Interestingly, CD44 co-localizes with P-glycoprotein to defined plasma membrane lipid microdomains (lipid rafts). 49 Moreover, in drug resistant cell lines, CD44 and P-glycoprotein co-immunoprecipitate. 50 More provocatively, agents that interfere with the function of P-glycoprotein affect the membrane distribution of CD44 and interfere with various CD44-dependent activities including cell motility and invasion. 50 Further emphasizing the complex nature of the relationship between HA production and both drug resistance and the development and pathogenesis of solid tumors, there is also evidence that certain ABC transporter proteins involved in multi-drug resistance may also play a role in the export of HA from cells.51,52 ABC-C (MRP5) appears to be particularly important in this regard.51,52 As one might expect, blocking the activity of MRP5 with the specific cGMP phosphodiesterase inhibitor zaprinast not only enhanced drug sensitivity but also reduced the migration and metastasis of human melanoma xenografts in SCID mice. 53
In addition to its differential expression within the tumor microenvironment and involvement in tumor development and pathogenesis, the physiochemical nature of HA makes it an attractive target for the therapeutic approach described in this study. Specifically, triggering of Fas-induced apoptosis is critically dependent upon receptor oligomerization and the natural ligands that bind to and activate Fas and other members of the TNF-receptor superfamily, including FasL, are all trimers.54,55 HA is a simple molecule composed of 2,000–25,000 repeating disaccharides of glucuronic acid and N-acetyl-glycosamine forming a linear structure with a molecule weight ranging from 105–-107 Da that adopts a pseudo random coil configuration in aqueous solution. 34 Studies suggest that HA oligomers containing 6–18 sugar residues bind to CD44 in a monovalent fashion while larger polymers are multivalent, have a higher affinity and efficiently crosslink CD44 inducing aggregation of the molecule in the plane of the plasma membrane.56,57 Since the minimum motif recognized by CD44 is repeated many thousands of times within a large molecular weight HA molecule, it was hypothesized that HA would be particularly efficient at binding to and crosslinking the CD44/Fas chimeras thereby inducing the degree of oligomerization necessary to trigger the desired apoptotic response. The results of the present study confirm that this is indeed the case. With regard to the safety of the proposed therapeutic approach, it is reassuring that both CD44/FasTM and CD44TM/Fas exhibit minimum toxicity in the absence of HA (Fig. 2) and only induce significant levels of cell death when it is added (Fig. 5).
For the CD44/Fas chimeras to be effective in cancer gene therapy, it is necessary not only that the corresponding ligand be present but that the receptor is active in the desired cell type. Differential activation of ligand binding within the tumor microenvironment would clearly be an advantage with respect to targeting specificity. In this regard, it is well established that the activation status of CD44 varies depending on the cellular context in which the molecule is expressed. Thus, while resting hemopoietic cells are mostly CD44-positive, they do not bind HA unless appropriately stimulated.58–61 Fortunately, when present on tumor cells, CD44 is generally constitutively active.59,62 While the mechanisms that regulate the affinity of CD44 for HA remain to be fully elucidated, various postranslational modifications including glycosylation,63–68 sulfation,69,70 phosphorylation71,72 and acylation 73 have been implicated in the process. The level of CD44 expressed by a cell is also clearly important. In keeping with this suggestion, the data shown in Figure 4 indicates that in transfected K562 cells, HA is only bound when CD44 or the CD44/Fas chimeras are expressed above a certain critical threshold.
In cells expressing a particular level of CD44, avidity for ligand can be further increased by clustering, which produces locally high concentrations of the receptor. This conclusion is supported by studies in which HA-binding can be induced by antibody-mediated crosslinking of CD44.64,74 There is also evidence that CD44 can form covalently linked homodimers following appropriate stimulation. 75 As evidenced by the Western blot data shown in Figure 3, spontaneous dimerization of wild-type CD44 clearly occurs in transduced 293 cells. Although it has been suggested that covalent dimerization of CD44 in lymphoid cells is critically dependent upon a cysteine residue (Cys286) present in the membrane spanning domain of the molecule,75,76 the fact that neither CD44TM/Fas nor CD44/FasTM exhibited significant evidence of dimerization when expressed in 293 cells (Fig. 3) argues that the cytoplasmic domain of CD44 may be more important, at least in this particular cell line. The limited dimerization of CD44 that is seen in untransfected 293 cells is in keeping with previous data suggesting that a certain threshold level of expression must be reached in order for CD44 to dimerize and/or form clusters. 77 Thus some stimuli that induce CD44 clustering and activation of HA-binding may do so simply by upregulating overall CD44 expression. 77 In any event, dimerization of CD44 is obviously not essential for HA binding as both CD44/Fas transgenes clearly possess this activity if expressed at a high enough level.
Although far less effective than the corresponding CD44/Fas chimeras, the results shown in Figure 6 demonstrate that CD44H alone can inhibit the survival and/or proliferation of tumor cells when introduced and expressed at a high level. Despite the fact that the interaction between CD44 and HA clearly promotes the proliferation of many diverse tumor cell types, there is much evidence that in some circumstances growth inhibition can result. Crosslinking of CD44 has, for example, been shown to induce a number of myeloid cell lines to arrest in G0/G1 and undergo terminal differentiation. 78 This effect appears to involve reduced expression and phosphorylation of c-jun, increased expression of p21 and reduced phosphorylation of Rb with a resultant decrease in the kinase activity of Cdk2 and Cdk4.79,80 Crosslinking of CD44 by immobilized antibodies was similarly shown to enhance the apoptotic response of lymphomas treated with dexamethasone. 81 In this case, the mechanism involves both downregulation of the anti-apoptotic protein Bclx(L) and upregulation of the pro-apoptotic protein Bax. 81 With regard to primary cells, ligation of CD44 can cause both dendritic cells and neutrophils to undergo apoptosis.82,83 Interestingly, the effect on dendritic cells appears to be mediated, at least in part, via HA-stimulated production of inducible nitric oxide synthase (iNOS). 82 Thus, while no clear consensus has emerged as to how CD44 can transduce both growth promoting and growth inhibiting signals upon antibody-mediated crosslinking or binding of ligand, the findings of these and other studies suggest that both the level of CD44 expressed by a cell, as well as the presence of particular alternatively spliced CD44 isoforms, are likely to prove important. Cellular context is also key, reflecting the expression and functional activity of various molecules that interact directly or indirectly with CD44 and/or transduce signals mediated via the receptor. Suffice to say, that until the mechanisms controlling the growth promoting and growth inhibiting activity of CD44 are better understood, the use of the wild-type molecule in cancer gene therapy is likely to prove problematic.
A potential problem with using Fas-induced apoptosis as an effector mechanism in cancer gene therapy is that defects in apoptosis are not uncommon in malignant cells.84–89 Fortunately, these are normally relative rather than absolute. Thus, while certain tumor cell lines, including the bladder line T24 used in this study, are resistant to conventional Fas agonists, such as the anti-Fas mAb C-11, apoptotic death could be induced upon infection with an adenoviral vector encoding FasL. 90 Clearly, the magnitude of the stimulus is a key component and resistance can be overcome if the trigger used is sufficiently active or present at a high enough level.
In summary, chimeric receptor proteins of the type described in this study provide a potentially powerful means of exploiting the differential presence of various multivalent ligands within the tumor microenvironment in order to induce targeted tumor cell death. Since the signals that activate the receptor have a restricted distribution, off-target effects are likely to be minimized as, even if the transgene is inappropriately expressed in non-targeted tissues, a response will not occur unless the required triggering signal is also present. That a certain threshold level of ligand is needed for apoptosis to be induced is another inherent advantage of this particular system allowing some “leakiness” in the activity the transcriptional or other elements used to target gene expression to tumor cells to be tolerated. As efficient induction of tumor cell death will only occur in tissue sites where both the chimeric receptor and corresponding ligand are highly expressed and active, it is hoped that systemic toxicity will be minimized. In vivo studies to test this hypothesis are currently underway.
Although early results are encouraging, it evident that a potential limitation of the pro-apoptotic gene therapy approach described herein is that only cells that directly take up and express the chimeric construct will be killed. Since it is unlikely that significant bystander killing will occur, success is thus critically dependent upon achieving efficient in vivo gene transfer. It is for this reason that we have chosen to focus our efforts on targeting, not tumor cells themselves, but instead the vascular endothelial cells that line the blood vessels upon which the survival and growth of a tumor depends. 91 The vasculature is an attractive target as even limited damage to a tumor-associated blood vessel may result in occlusion and the cessation of blood flow thereby preventing the delivery of oxygen and nutrients to many thousands of tumor cells. It thus follows that even the relatively poor transduction efficiencies that can realistically be obtained using currently available delivery systems is likely to translate into greatly amplified “bystander” killing of malignant cells. Results obtained with small molecule “vascular disrupting agents” (VDA) such as Combretastatin A4 and ZD6126 serve to validate this strategy.92–95
Disclosures
The authors report no conflicts of interest.
Footnotes
Acknowledgements
These studies were supported by grants from the National Cancer Institute (CA100004), the Arizona Biomedical Research Commission (Contract 0706) and the University of Arizona, Women's Cancers Better Than Ever Program. We gratefully acknowledge the technical support and other input of Dr. Carmine Carpenito.