
Abstract
Despite the considerable progress in understanding the biology of human cancer and technological advancement in drug discovery, treatment failure remains an inevitable outcome for most cancer patients with advanced diseases, including melanoma. Despite FDA-approved BRAF-targeted therapies for advanced stage melanoma showed a great deal of promise, development of rapid resistance limits the success. Hence, the overall success rate of melanoma therapy still remains to be one of the worst compared to other malignancies. Advancement of next-generation sequencing technology allowed better identification of alterations that trigger melanoma development. As development of successful therapies strongly depends on clinically relevant preclinical models, together with the new findings, more advanced melanoma models have been generated. In this article, besides traditional mouse models of melanoma, we will discuss recent ones, such as patient-derived tumor xenografts, topically inducible BRAF mouse model and RCAS/TVA-based model, and their advantages as well as limitations. Although mouse models of melanoma are often criticized as poor predictors of whether an experimental drug would be an effective treatment, development of new and more relevant models could circumvent this problem in the near future.
Keywords
Introduction
Melanoma is the malignancy of the pigment-producing melanocytes in the skin and characterized by its highly aggressive nature to metastasize to distant organs. Despite recent advances in melanoma therapeutics, prognosis remains poor, with a five-year survival rate of 16% for patients with distant metastases. 1 Furthermore, the incidence of malignant melanoma continues to rise year-over-year, with almost 74,000 new cases in the United States projected for 2015. As such, melanoma is a significant health issue. Nevertheless, some research efforts have resulted in a variety of emerging treatment modalities that offer improved patient outcomes against the deadly disease. In 2011, the US Food and Drug Administration (FDA) approved vemurafenib (Zelboraf®), a mutant BRAF inhibitor for advanced stage melanoma. 2 Unfortunately, while BRAF-targeted drugs are effective at short term in inhibiting tumor growth and progression, their effects are not persistent, and recurrence of metastatic disease is common. Reports suggest that multiple mechanisms can lead to development of resistance to BRAF-targeted therapies.3,4 Combining BRAF and mitogen-activated protein kinase kinase (MEK) inhibitors results in better outcomes than single agents, but resistance, although delayed, remains inevitable.5,6 Relapse is attributed to the reactivation or overactivation of MEK, PDGFR, RAS, COT, or AKT pathways and is further complicated by the presence of heterogeneous populations of tumor stem cells.3,4,7
Immunotherapy is another exciting treatment strategy that has seen comparable clinical successes. 8 Two FDA-approved agents, ipilimumab (Yervoy®) and pembrolizumab, target immune-modulatory proteins (cytotoxic T lymphocyte-associated antigen 4 [CTLA-4] and programmed cell death protein 1 receptors, respectively) and result in upregulation of host immune response against melanoma by blocking negative T-cell regulators.9,10 In phase III trials, ipilimumab and related immune therapies have been found to produce improved overall survival. 8 However, these agents have also experienced their own challenges. Response rates, ~10%–15%, have been less than ideal, and both relapse and immune-related side effects have been reported, the mechanisms of which are not wholly understood. 8 Ultimately, the development of new regimens that would resolve the shortcomings of existing solutions will require a broader understanding of tumor biology. This can be achieved through the use of in vivo models that accurately simulate true melanoma behavior. Indeed, cancer cells exist as diverse entities surrounded by a microenvironment that includes blood vessels, extracellular matrix, and host immune cells, each of which play critical roles in the response of tumors to therapeutic agents. Models must not only recreate these features but also recapitulate natural tumor progression, from proliferation to invasion and metastasis. Representation of all these facets will facilitate and advance translation of discoveries to the clinical setting and ensure rapid delivery of new treatments to melanoma patients.
The most widely used preclinical model that employs the aforementioned characteristics is the murine model. They owe their ubiquity to their ease of manipulation and availability as well as existing knowledge base regarding their genetics. A number of melanoma murine models are used, including xenograft, syngeneic, and genetically engineered models.11–18 Xenograft models involve the culturing and engraftment of human melanoma cells into immune-deficient mice. They can be easily established and manipulated to quickly identify key pathways responsible for malignancy. Syngeneic xenograft transplantation involves the induction and transplantation of melanoma cells into same species and genetic background.19,20 As animals can possess a functional immune system in this model, syngeneic transplantations are frequently used to evaluate immunotherapies and interactions between cancer and immune cells, such as dendritic cells. 21 Genetically engineered models (GEMs) use transgenic mice with modified gene expression to determine the mechanisms of melanomagenesis. These models have been useful in elucidating gene function and identifying key targets for therapeutics.22,23 They have been combined with other modes of tumor induction, such as by ultraviolet (UV) or carcinogens, to determine predispositions and risk factors of melanoma development. 1 Each model reveals unique insights into melanoma behavior, and given their distinct advantages and disadvantages, it is necessary to utilize all three to improve our understanding of the disease and drive treatment development.
Xenograft Transplantation Models
The development of xenograft models was a big step in moving toward more clinically relevant tumor models.24,25 Since the first report of the successful xenografting of a human cancer cell line into nude mice in 1969, numerous studies have been conducted using the xenograft mouse model as a tool to answer a variety of questions regarding the cause, prevention, and therapy of various malignancies, including melanoma.26,27 In recent years, human tumor xenograft models have been used widely to evaluate the targeted therapies27,28 and to test the combinatorial efficacy of therapeutic agents.11,29,30
Cell line xenografts
Human tumor xenografts implanted subcutaneously (SC) into immunosuppressed mice have played a significant role in cancer drug discovery for the past 25 years.11,24,25,29–32 This model employs the SC transplantation of established human melanoma cell lines into immunocompromised mice that do not reject human cells. For example, nude athymic (nu/nu) mice that are T-cell deficient, 33 or severe combined immune-deficient (SCID/SCID) mice that are deficient in both T-cells and B-cells, 34 are routinely used for propagating human cell lines SC (cell line xenografts) to reconstitute solid tumors. The advantage of this model is that it allows human melanoma cells to directly establish interactions with the lymphatic and blood vessels to study the tumor growth behavior and drug response in vivo.35–37 Further, use of similar culture conditions of injected cancer cells (eg, number and passage) helps control over the timing of tumor growth enabling to generate data that are easily comparable. For metastatic melanoma cell line xenografts, cells are often injected SC and infrequently intradermally. Although intradermal injections mimic the primary melanoma better because of tumor formation, mouse skin becomes ulcerated in a short period of time and early termination of the experiment is required.
SC xenografts can be useful to study metastases. Many metastatic melanoma cell lines have been reported to metastasize spontaneously from the primary xenografts to distant sites such as the lung.35,38,39 However, not all metastatic cells injected SC have the ability to metastasize. For example, human variant melanoma cell lines, 1205 Lu and 451 Lu, were selected from the lung of a nude mouse after several in vivo passages of WM164 and WM793B melanoma cells isolated from the metastasis of a melanoma patient. The WM164 or WM793B cells were not competent for metastasis in nude mice prior to this selection. These studies show that metastases are produced by the selective growth of specialized aggressive subpopulations of metastatic cells that preexisted in the parent tumor.40,41 Another approach is experimental metastases in which tumor cells are injected into the tail vein or retro-orbital vein to form metastatic lesions in the lungs. While this approach is frequently used as a metastasis model, the drawback is that it bypasses actual events of metastasis in a patient scenario.35,36,38,39,42,43
Cancer cell lines are the most widely used starting material for xenograft models, as they are easily available and easily propagated in immune-deficient mice for in vivo tumor growth. However, most of the melanoma cell lines have been established under nonphysiological conditions for several years resulting in selection of clones that differ significantly from the originating cells and are no longer representative of the original tumor.
Hence, cell line xenograft models are often poorly predictive of clinical outcome, and drugs showing efficacy in this model often fail in clinical trials. 44 The use of primary melanoma cells for xenografting has also been reported. 45 Primary melanoma cells were tittered down to single cells to generate xenografts in NOD/SCID IL-2 receptor gamma chain knockout (NSG) mice. 45 Whether melanoma xenografts use established cell lines or primary tumor cells, owing to disadvantages mentioned earlier, and the fact that xenografts do not grow in their natural tissue and have inadequate tumor microenvironments that include the lack of an immune system, better models are required.
Patient-derived tumor xenografts
In an effort to address the aforementioned concerns, there has been a recent increase in the use of patient-derived tumor xenografts (PDTXs) engrafted into immunocompromised rodents such as athymic nude or NSG mice. The history of utilizing PDTX models in drug discovery can be traced back to several decades ago. One of the earliest reports was by Fiebig et al, 46 whose group demonstrated 92% accuracy in predicting the efficacy of number of chemotherapy drugs at their respective maximal tolerated doses (MTDs) and 97% in predicting no-response in PDTX models derived from 34 patients. 47
Recently, as preclinical models, PDTXs are being characterized and gaining popularity at some point during the preclinical discovery and translational research stages because of significant increases in their availability and affordability.48,49 Collecting evidence has now shown that PDTX models are superior to traditional cell line xenografts, as they maintain more similarities to the tumors found in actual patients; accurately reflect human cancer and provide a practical solution by preserving the fidelity of clinical characteristics; and provide tumor supply for drug discovery, target identification, and validation strategies.48,50,51 PDTX models may more accurately reflect clinical response when treated with therapeutic agents at clinically relevant doses. 52 Several studies have shown strong conformity in histology, transcriptome, polymorphism, and copy number variations between PDTX models and parent tumors. Strong preservation of the chromosomal architecture was observed.53–56
Numerous tumor-specific PDTX models have been established, including melanoma.57,58 Using patient-derived tumor grafts, it is now possible to study the responses of metastatic melanomas within in vivo three-dimensional environments. These models are now being used for the identification of drug resistance switches and combination therapy regimens that prevent drug resistance as well as the modeling of emergence of drug resistance. For example, recently, a BRAFT1799A mutated melanoma model called HMEX1906 was developed by using an early passage, vemurafenib-naive, primary human-patient-derived xenograft.47,59 To generate drug-resistant melanomas, tumor-bearing immunocompromised mice were dosed for eight weeks with 45 mg/kg body weight of vemurafenib. This resulted in >80% inhibition of phosphorylated ERK1 and ERK2 for up to 24 hours, which was similar to the degree of inhibition observed in clinical trials.47,59 However, 56 days after dosing was started, in two out of 10 mice, emergence of drug resistance was overserved. Fragments of one of the resistance tumors were harvested, reimplanted into a new group of mice, and treated with 45 mg/kg vemurafenib to generate drug-resistant tumors for determining the mechanisms of resistance. 58 There are clear advantages of using these models. As discussed earlier, PDTX models are heterogeneous in nature and more closely reflective of tumors in actual patients. 60 It permits the sampling of serial biopsies from a single tumor and dozens of mice can be created from a single patient sample in order to investigate the presence of more than one clonally derived mechanism of resistance within the original tumor. 58 A large collection of PDTX models can best represent a comprehensive patient population with different preexisting mutations and susceptibility to generate additional mutations. Another major advantage of using PDTX for target identification and validation is that the process from target identification to validation and then to efficacy screening can be rationalized around the same models, hence, offering a complete circle from the patient to the mouse and then back to the patient.
More recently, concept of PDTX mouse clinical trial is evolving and has already started to yield positive results that are helpful in guiding clinical management of the patient's tumor. 61 For this, PDTX models established from the very same patients on trial are being treated ahead of patient therapy or concurrently, and results from the mouse trial are provided in real time. Further powered by the molecular characterization of the tumors, this highly personalized approach has the potential to revolutionize the drug development and patient care. 62 A potential use of PDTX models in personalized therapy of melanoma is depicted in Figure 1.
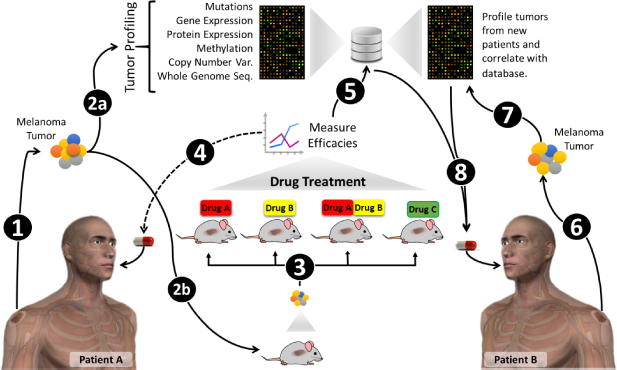
Schematic showing potential use of PDTX models in personalized therapy of melanoma. Tumors surgically removed from patients (1) are profiled in multiple platforms (2a) and also transplanted into mice for development of PDTX model (2b). PDTX model is serially propagated to test various therapeutics or their combinations (3). Based on the response of PDTX model, best therapeutic strategy is selected for the patient (4). The tumor profiles and PDTX efficacy data would be stored in the databases (5) and could provide a useful tool for the selection of therapeutic strategy for new patients (6–8).
Although PDTX models present an exciting opportunity for improving predictive value of preclinical and translational studies and indicate several advantages over conventional cell line xenograft models, just like any other preclinical models, some key challenges that remain before this strategy can be broadly implemented in clinical practice. First, establishment of PDTX models is still a technically challenging and time-consuming process. Basically to establish a PDTX model with human melanoma specimens, following surgical removal, the viable tumor is dissected into small pieces and directly transplanted SC into nude mice. Time for palpable tumor to develop typically ranges from three to nine months, and in many cases, tumors fail to develop. Moreover, xenografts from primary melanomas take even longer. Second, this model uses severely immunocompromised host mouse strains, mainly the nonobese diabetic SCID gamma mice, despite the fact that this strain allows higher intake rate and consistent growth of xenografted human tumors, which do not grow in the context of an intact immune system, thus fail in modeling immune responses. Even though patient tumor biopsy containing human stroma components including immune cells, which can be grafted together with the tumor tissue, 63 they usually cannot survive beyond the first passage, and in the subsequent expansion, they completely get lost. 64 Furthermore, the lack of functional immune system limits the utility of these models in studies where immune responses are required; hence, immunotherapy cannot be readily studied in this model. It is well documented and accepted that immune system is an important part of tumor stroma and significantly contributes to tumor initiation, progression, and metastasis, as well as therapeutic response. 65 Mice with partially or completely humanized immune systems can potentially overcome this issue; however, still significant technical challenges exist. 66 Unfortunately, introduction of human fibroblasts and vasculature is rapidly replaced by murine counterparts. 64
Furthermore, PDTX models are difficult to manipulate genetically compared to cell lines. Most PDTX models are established from and passaged as tumor fragments, and conventional transfection or transduction are not efficient to genetically modify the tumors or introduce detection markers such as luciferase or fluorescent proteins. 67 Additionally, despite technical advances that have increasingly improved the tumor take, success rate still varies because of different tumor types and different subtypes within the same tumor type. Even though using PDTX models' artificial selection in extended culture on plastic can be avoided, the in vivo selection process does exist as soon as the tumors are implanted. For instance, high-grade, fast proliferating tumors tend to be easier to establish as PDTX models than low-grade, slowly growing but progressive tumors. 68 In conclusion, to effectively demonstrate the feasibility and clinical benefit of the PDTX-guided treatment prioritization in the patient care setting, properly controlled clinical trials are needed.
Syngeneic Transplantation Models
Syngeneic transplantation models date back to the 1950s and have been recognized as a useful platform for studying melanoma behavior and metastasis. 69 These models have important benefits over those that use immunocompromised mice as they allow for the interaction of melanoma cells with competent T-cells and B-cells found naturally in the human melanoma microenvironment. 17
Syngeneic models employing the B16 cell line
Several cell lines have been used for syngeneic transplantations, including Harding-Passey cells isolated from the ICR mouse ear and the Cloudman S91cell line obtained from the DBA mouse. 69 The most widely used cell type, though, has been the B16 cell line that spontaneously forms tumor after chemical induction of melanoma in C57BL/6J mice and gives rise to a diverse spectrum of subclones with various propensities for proliferation, invasion, and metastasis. Two well-established subclones of the B16 melanoma cell line, obtained from in vivo passaging, include the B16F1 and B16F10 variants. B16F1 is characterized by low metastatic potential and, thus, is useful for studying primary tumor growth. 69 Conversely, B16F10 has high metastatic potential to distant visceral organs, most notably the lungs, and has been ideal for in vivo studies because of its swift growth pattern and high turnover, inducing death within two to four weeks after SC injection into mice. 70
The B16 cell line is a frequently used model to study the efficacy of various treatments, especially immune therapies that harness the available T-cells to target melanoma-specific antigens in host mice. B16 cells express low levels of major histocompatibility complex class I markers and are, thus, poorly immunogenic because of their inability to be recognized by cytotoxic CD8+ T-cells. 71 While this has impeded some efforts to develop vaccine-based therapies, it has been found that B16 cells possess high levels of other targetable epitopes associated with melanoma, including gp100 and tyrosinase related protein 2 (TRP2). 17 It has been demonstrated that vaccination of mice with the Trp2 epitope elicited an immune response that inhibited B16F10 growth and protected mice from lung metastasis and partially from SC tumor formation. 72 Adjuvant therapy with high-dose IL-2 has been found to enhance the Trp2 vaccine's anticancer effects. 71 Various other cytokines, including TNF-α, have been used with similar success to prolong survival of tumor-bearing mice.
An obvious disadvantage of B16F10 syngeneic transplantation models is the use of murine cell lines that possess differences in adhesion proteins and growth factor production compared to human melanoma.
70
Although B16 cells can give rise to various subclones, they originate from a single, inbred strain of mice and are not representative of the genetic diversity of human cells.
73
Furthermore, the enzymes and mechanisms used for its invasion into host tissues and downregulation of its apoptotic cascade differ from their human counterparts. Analysis of the B16 mutanome has revealed inactivating mutations in
Other models
Other cell lines have been employed in syngeneic transplantation models. Harding-Passey cells are obtained from the dermal melanoma of ICR mice and BALB/c × DBA/2F1 mice and have been used to induce intracranial tumors. 69 While they cannot metastasize spontaneously, their ability to produce melanin has been particularly useful for studies involving the effects of melanin content on the metabolic function of melanoma. The Cloudman S91 cell line, isolated from DBA/2 mice, has been used to assess the efficacy of several novel anticancer therapies and drug delivery modalities, including targeted polyethylenimine polyplexes and nanotransporters.76,77
Genetically Engineered Mouse Models
Without doubt, understanding the genetic alterations that trigger melanoma development is an indispensable step for the development of novel melanoma therapeutics. Genetically engineered mouse models (GEMMs) have been extensively used to investigate the effect of genetic alterations in melanoma initiation, progression, and metastases (Table 1).22,23 In contrast to other preclinical models, GEMMs are suggested to be a more accurate predictor for drug efficacy assessment. 23 Despite the great use of transgenic animal models, several limitations exist. Multiple mouse strains need to be interbred in a costly and labor-intensive manner. In many cases, tumors in multiple tissues can arise, limiting the use of the generated model. Furthermore, some of the genetic alterations have deleterious effects on reproductive fitness; hence, obtaining the desired genotypes would not be possible. However, advancements in molecular biology and genetics led to the development of state-of-the-art tissue-targeted inducible expression systems and have helped to overcome many of the aforementioned limitations. In the following section together with state-of-the-art melanoma models, various GEMMs of melanoma are discussed.
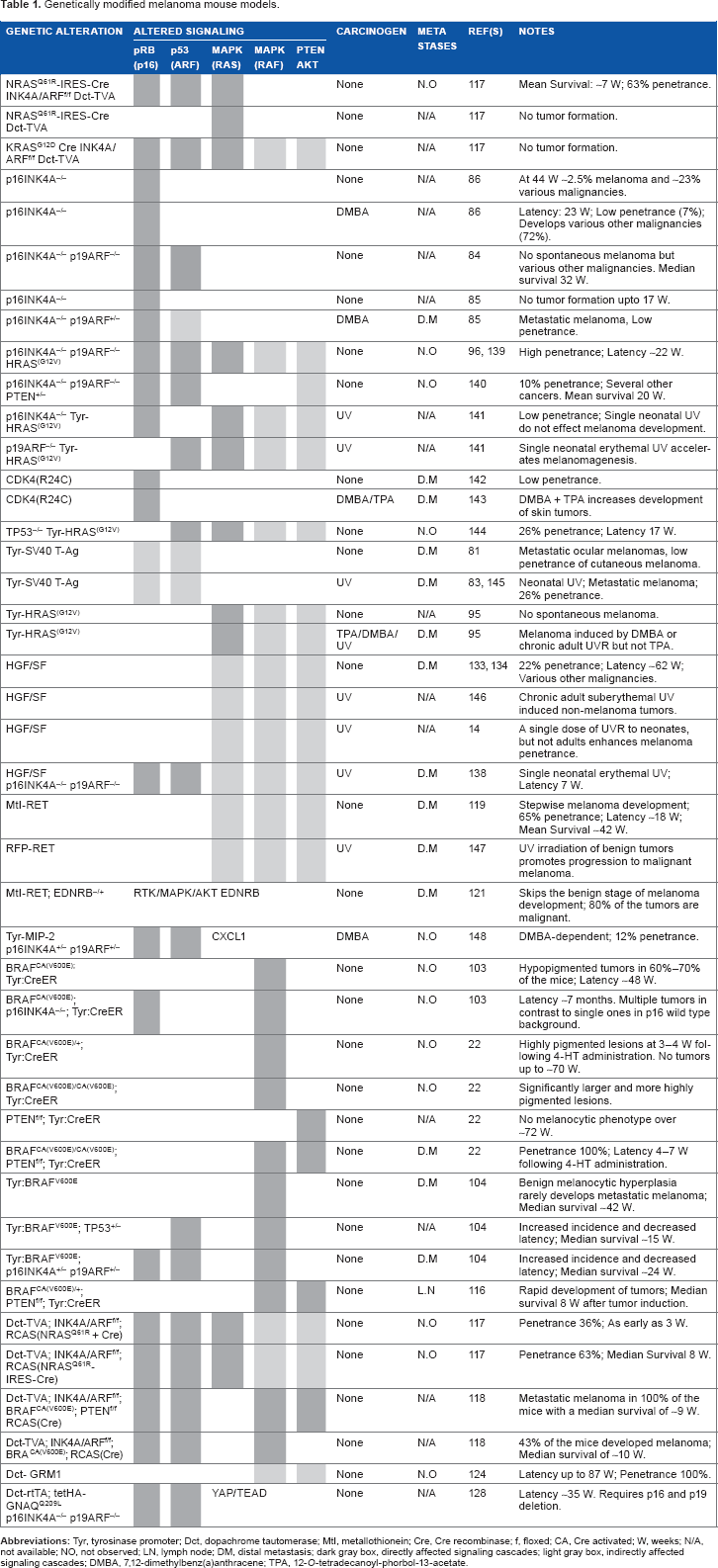
Genetically modified melanoma mouse models.
CDKN2A models
Earliest findings regarding the genetics of melanoma were obtained from the linkage studies of familial melanoma cases.78,79 Cyclin-dependent kinase inhibitor 2A (CDKN2A) locus located at 9p21 was identified as a melanoma susceptibility region and encodes two well-identified tumor suppressor proteins, p16INK4A and p14ARF (p19ARF in mouse).
80
Although these proteins are structurally unrelated, they are expressed from the alternative reading frames of a common gene (
In 1991, Bradl et al generated one of the first genetically engineered mouse melanoma models via tyrosinase promoter driven, melanocyte-specific expression of simian virus 40 (SV40) T-antigen. 81 Expression of this viral protein was analogous to the loss of CDKN2A locus, as it simultaneously disrupts pRB and p53 signaling cascades. 82 However, in this model, melanomas were predominantly originating in the eyes, and skin melanomas were infrequent and mostly benign. In 1994, Klein-Szanto et al incorporated short-term UV irradiation into this model and reported higher incidence of cutaneous melanoma without other skin tumors. 83 In 1996, Serrano et al developed a mouse strain with targeted deletion of CDKN2A locus. 84 These animals developed various malignancies but not melanoma. Later on, several studies suggested that loss of Ink4a or Arf was not enough to trigger melanoma development but makes animals susceptible to UVR or carcinogen-induced melanomagenesis.85–87
RAS models
In the late 1980s, RAS family of proteins was discovered to be frequently mutated in cutaneous melanoma.88–91 About 15%–25% of human melanomas harbor activating mutations in NRAS, whereas Harvey rat sarcoma viral oncogene homolog (HRAS) and Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations are very rare (<2%).92–94 The importance of RAS mutations in melanoma development was studied in vivo through their melanocyte-specific expression in mice.95–98 Tyrosinase-driven expression of activated HRASV12G was not able to trigger spontaneous melanoma development.95,99 However, in these animals, either UVR or 7,12-dimethylbenz[a] anthracene (DMBA) treatment was able to induce melanoma development in a relatively short latency (57% at 52 weeks with UVR; >80% at 45 weeks with DMBA).95,99 Interestingly, p16INK4a was found to be deleted in the developing tumors, and cross-breeding HRASV12G mice with p16INK4a/p19ARF knockout mice led to a new model with the capability of developing a large number of spontaneous cutaneous melanomas with a shorter latency (60% at 26 weeks). 96 It was subsequently shown through a doxycycline-inducible HRASV12G version of this model that the developed tumors were strictly dependent on HRAS activity since downregulation of HRAS via withdrawal of doxycycline led to their total regression. 95 On the other hand, melanocyte-specific expression of NRASQ61K was able to trigger melanoma development with a low incidence rate and high latency (29% at 70 weeks). 97 Cross-breeding this mice with p16INK4a/p19ARF knockout mice increased the incidence rates, while decreasing the latency (83% at 65 weeks for INK4a+/– mice and 94% at 42 weeks for INK4a+/– mice). The developed tumors were aggressive and >30% of the animals had metastases into lymph nodes and distant organs. 97 Another NRAS mutation (NRASG12D) was assessed for its potential to induce melanomagenesis when expressed in the melanocytes of developing mouse embryos. 100 Despite this mutation was able to induce melanocytic lesions resembling blue nevi, it failed to induce spontaneous cutaneous melanoma up to 24 months. However, it triggered development of primary melanoma in the central nervous system (CNS) of the animals. Although NRAS is rarely involved in primary melanoma of CNS, it is suggested that this model could represent some of the rare cases. 101
PTEN/BRAF models
Identification of the PTEN deletion and BRAF mutations were two of the greatest discoveries in melanoma field. In 2002, as an early product of Cancer Genome Project, BRAF was discovered to carry somatic mis-sense mutations in >65% of malignant melanomas. 102 BRAF mutations are mutually exclusive to RAS mutations, and taken together, the mitogen-activated protein kinase (MAPK) signaling is overactivated in more than ~85% of the malignant melanoma cases. Melanocyte-specific expression of BRAFV600E protein consistently leads to benign melanocytic lesions. Although some studies reported induction of melanoma (~54% at 12 months), others were controversial to these observations and did not report melanoma development.22,103,104 However, loss of p16INK4a or p16INK4A/p19ARF decreases the latency and increases the tumor penetrance (~80% at 12 months).103,104 It has been shown that ~80% of the benign nevi carry BRAF V600E mutation, suggesting that BRAF mutation is not sufficient to drive melanoma development and requires additional alterations. 105 In fact, virtually all the tumors that were developed in BRAF V600E mice had increased AKT activity and decreased p16INK4A expression. 104
Frequent loss of heterozygosity of chromosome 10q23 in melanoma led to the identification of PTEN phosphatase as a tumor suppressor protein that is deleted in ~20% of uncultured primary and 55% of metastatic melanoma tumors.106–110 Deletion of PTEN leads to overactivation of AKT signaling and frequently accompanies BRAF activation.111–113 In 2006, two different groups generated melanocyte-specific inducible Cre recombinase (Tyr:Cre-ERT2) transgenic mice.114,115 These mice were used to generate state-of-the-art melanoma mouse models to investigate BRAF V600E-induced melanoma and its interaction with PTEN deletion.22,116 In these models, topical application of tamoxifen triggers melanocyte-specific expression of Cre recombinase to knockout floxed PTEN gene and/or activate the mutant BRAF protein expression. In 2009, using this model (Tyr:Cre-ERT2 BrafCA/+), Dankort et al reported that melanocyte-specific expression of V600E BRAF triggers formation of highly pigmented lesions in 21–28 days following topical tamoxifen application. These lesions did not progress to malignant melanoma up to 80 weeks. Tyr:CreER mice that were homozygous for catalytically active BRAF (Tyr:Cre-ERT2 BrafCA/CA) developed larger and more highly pigmented than heterozygous ones indicating the dosage effect of BRAF on melanocyte proliferation. On the other hand, melanocyte-specific knockouts of PTEN (Tyr:CreER; PTENlox/lox) did not elicit any melanocytic lesion up to 18 months. However, combination of PTEN knockout with mutant BRAF activation (Tyr:Cre-ERT2 BrafCA/+; PTENlox/lox) significantly enhances melanocytic lesion development as early as 7–10 days following tamoxifen application. The cooperation between mutant BRAF and PTEN silencing was so strong that all mice rapidly developed advanced metastatic malignancy that required euthanasia in 25–50 days. Metastases to regional lymph nodes and lungs were observed. Similar results were obtained when the same genetic modifications were performed to C57BL/6J mouse strain. 116
RCAS/TVA system
More recently, with the advancements in molecular biology and retroviral-vector delivery systems, a new technique called Replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor/tumor virus A (RCAS/TVA) system has been developed to create animal models of diseases.17,18 This system uses a replication-competent avian retrovirus Replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor/tumor virus A vector to induce delivery of cDNAs, short hairpin RNAs, microRNAs, and other noncoding RNAs in an efficient, stable, and targeted manner. 18 Targeting to specific cells is established through expression of tva950 or tva800 proteins. Mammalian cells lack these proteins, hence are normally resistant to infection by RCAS virus. Ectopic expression of TVA in specific cell types or tissues renders their susceptibility to infection by RCAS. This system offers unique advantages in contrast to conventional mouse models of cancer. 17 First of all, multiple genetic alterations can be introduced rapidly and in a sequential manner without the requirement of crossing multiple mice strains. Hence, RCAS/TVA system could allow rapid assessment of newly identified genes on disease progression and maintenance. Second, in this model, tumor microenvironment is mimicked better as cancer develops from few modified cells that are surrounded by normal cells. Furthermore, combination of this system with various genetic engineering approaches, such as Cre-Lox recombination system and Tet-on/Tet-off system, greatly enhances the opportunities for development of new mouse models. For instance, RCAS-mediated delivery of Cre recombinase to targeted cells that contain “floxed” genes eliminates the need to express Cre recombinase from tissue-specific and/or inducible promoters, hence preventing leaky expression problems. However, as each model has its own advantages and disadvantages, RCAS/TVA system has few limitations. It requires actively dividing cells, and integration is thought to be random, potentially affecting the expression of host genes. Genes that are > 3 kb cannot be successfully delivered.
VanBrocklin et al used the RCAS/TVA system to assess the NRASQ61R-driven melanoma in INK4A/ARFlox/lox mice. 117 Transgenic mice expressing TV from the dopachrome tautomerase (DCT) promoter were crossed with INK4A/ARFlox/lox mice to generate Dct-TVA; INK4A/ARFlox/lox mice. Administration of RCAS virus encoding NRASQ61R and Cre recombinase led to development of melanomas in 36% of the mice in as early as three weeks. Linking the expression of NRASQ61R and Cre by an internal ribosomal entry site (IRES) resulted in tumor formation in 63% of TVA-positive mice.
The same group crossed Dct-TVA; INK4A/ARFlox/lox mice with BRAFCA; ±PTENlox/lox mice that carry a Cre activated BRAFV600E floxed PTEN gene. 118 RCAS-mediated delivery of Cre recombinase to Dct-TVA; INK4A/ARFlox/lox; BRAFCA; PTENlox/lox mice resulted in metastatic melanoma formation in 100% of the mice with a median survival of 62 ± 6.7 days. In wild-type PTEN background (Dct-TVA; INK4A/ARFlox/lox; BRAFCA), 43% of the mice developed melanoma with a slightly increased median survival of 72 ± 6.7 days. However, importantly, no metastasis was observed in these mice.
RET model
Another mouse melanoma model was generated using RET proto-oncogene, a receptor tyrosine kinase for glial cell-derived neurotrophic factor (GDNF) family of signaling molecules.119,120 In this model, expression of RET under the control of ubiquitous metallothionein-1 promoter leads to melanoma development in a stepwise manner. Accordingly, no tumors are observed for several months after birth, which is followed by growth of multiple benign melanocytic tumors that eventually become malignant and metastasis to distant organs. During this progression, a gradual increase in the expression of RET transgene is observed, which is accompanied by activation of MAPK and-cJun signaling cascades.
It was discovered that melanomas arising from RET mice express decreased levels of endothelin receptor B protein (EDNRB) (an essential receptor for the development of neural crest-derived cell lineages, including melanocytes).
121
Crossbreeding RET mice with
GRM1 model
Pollock et al accidentally developed mouse model of melanoma that involves aberrant expression of metabotropic glutamate receptor 1 (GRM1). 124 Melanocyte-specific expression of GRM1 via Dct promoter was found to trigger development of spontaneous, highly pigmented melanomas in skin, eyes, and ear of the animals with 100% penetrance. In homozygous animals, the onset of the melanomas were -three months, while in heterozygous, they were first detectable -seven months. Aberrant GRM1 expression was discovered to contribute to some cases of melanoma. A single-nucleotide polymorphism in this gene was significantly associated with melanoma susceptibility in patients with a low level of sun exposure. 125 Furthermore, in contrast to no-expression in normal skin, −40% of melanoma samples were found to express GRM1 protein. 126 More recently, expression of GRM1 was reported in uveal melanoma, and this model was suggested to be used as a spontaneous uveal melanoma model. 127
GNAQ model
Another mouse model for a rare subtype of melanoma, which involves guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) mutation, was recently developed. 128 Although mutation of this gene was rarely observed in cutaneous melanoma (<2%), it is constitutively activated ~50% of primary uveal melanomas because of mutation of codon 209. 129 To investigate the downstream alterations mediated by GNAQ activation and to assess whether activated GNAQ can drive melanocyte transformation in vivo, Feng et al generated a mouse model expressing hemagglutinin A epitope tagged-mutated GNAQQ209L protein under the control of tet-responsive elements and crossed them with mice expressing the reverse tetracycline-activated transactivator (rtTA2) under the control of Dct promoter (Dct-rtTA). These animals were not able to develop melanoma up to 40 weeks. However, when they were crossed with mice defective in p16INK4A and p19ARF genes, >50% of the mice developed cutaneous melanoma with a latency of ~35 weeks, following doxycycline treatment. In the developing lesions, transcriptional coactivator, Yes-associated protein 1 (YAP1), was localized to nucleus to drive oncogenesis via association with the TEA domain (TEAD) family of transcription factors. 129
Taken all together, many different melanoma mouse models have been established to investigate the significance of melanoma driving genetic mutations. Models involving deletion of CDKN2A locus, TP53 and PTEN genes; activation of GNAQ, GRM1, stem cell factor (SCF), hepatocyte growth factor/scatter factor (HGF/SF), Met, RAS, and most importantly, BRAF have been developed. However, new mutations are being identified with the advancement of next-generation sequencing technology. 130 Therefore, we hope that in the near future, newer and more advanced melanoma models will be developed.
Induction with Physical Agents or Chemically Induced Models
UVR-induced models
Models that involve the induction of melanomagenesis through UVR treatment can be useful for simulating the natural progression of melanoma development as it occurs in human beings. However, the difference between localization of melanocytes within the mice and human skin is an important concern for mouse melanoma models and, especially, for UVR-induced ones. 71 Human melanocytes reside primarily in the basal layer of the epidermis and within the epidermal-dermal junction; hence, susceptible to penetration by UVR penetration. Mouse melanocytes, however, exist largely in the roots of hair follicles but rarely at the dermal-epidermal junctions. 72 While some melanocytes can also be found in the epidermis of hairless regions such as the ears, tail, and paws, melanoma rarely develops spontaneously in mice as the dermal melanocytes reside deeper in the skin and are better protected from UVR. 72 As such, melanoma development in mice presents with distinct histological properties, which are not representative of the human disease.17,122
In human melanoma progression, the transition of tumor from radial to vertical growth phase (RGP to VGP) is an important measure for clinical outcome and requires invasion of dermis by epidermal melanoma cells. 72 Owing to the absence of melanocytes from the epidermal–dermal junction, mouse melanoma models lack RGP to VGP progression. To overcome this problem, various approaches have been employed.131–133 Induction of keratinocyte-specific expression of SCF, the ligand for the kit receptor tyrosine kinase, has been shown to alter melanocyte localization leading to development of mice skin that more closely mimics human skin in contrast to normal mice. 131 Similarly, it has been shown that keratin 5 promoter-driven expression of endothelin 3, a ligand for EDNRB receptor, can compensate for Kit's role in early melanocyte maturation and lead to development of pigmented mice skin harboring dermal melanocytes. 132
An important melanoma mouse model with unique distribution of melanocytes has been created via overexpression of HGF/SF under the control of metallothionein gene promoter.133,134 HGF/SF provides important mitogenic and morphogenic cues to melanocytes and has been implicated in tumorigenesis through binding of the MET receptor tyrosine kinase and activation of the MAPK and PI3K pathways. 135 HGF/SF transgenic mice possess melanocytes in the epidermis, upper dermis, and epidermal-dermal junction akin to human melanocytes, and the development of metastases after UV irradiation follow human etiology: benign nevus, RGP, VGP, and late metastatic spread to visceral organs.133,135
Around 22% of the HGF/SF transgenic mice develop spontaneous melanomas with a mean onset of 15.6 months; however, these melanomas do not reflect the human melanomas as they demonstrate a dermal morphology. Exposure of neonatal animals to UV radiation results in the development of lesions resembling RGP/VGP melanoma and invasive melanoma with junctional and dermal components. 14
HGF/SF transgenic mice have been notably used to determine the band of UV most implicated in melanomagenesis. It was found that in neonatal HGF/SF mice, tumorigenesis occurred after ultraviolet B (UVB) irradiation but not after ultraviolet A (UVA) treatment. 69 Neither UVA nor UVB induced metastatic melanoma in the adult phase, and any tumors induced had long latency periods. These findings emphasize the significance of childhood UV exposure as a risk factor to melanoma development. The differences in the effects of UVA and UVB on mouse melanomagenesis can be attributed to the mechanisms with which they cause DNA damage. UVB has been found to be more carcinogenic because it can generate direct cyclobutane pyrimidine dimers (CPDs) and 6-4PP lesions that are not effectively repaired. 72 Both photoproducts cause damage by distorting the integrity of the DNA helix, preventing RNA polymerase from actively transcribing and resulting in gene inhibition. 136 The UVB signature mutations such as CT->T and CC->TT transitions may be used for identification of UVB-specific damage. Upon UVR stimulation, host cells generate a p53-mediated stress response that attempts to repair UVB signature mutations. 72 p53 seems to play a role in UV-related repair, and recent studies have indicated that TP53 becomes upregulated upon UV irradiation. Existence of additional mutations, such as BRAF (V600E), contributes to the development of nevi that indicate melanomagenesis. 137
Unlike UVB, UVA causes DNA damage indirectly by stimulating photosensitizers, such as melanin, to generate reactive oxygen species (ROS). 72 ROS-mediated oxidative stress triggers formation of oxidized bases such as 8-oxoguanine (8oG) and thymine glycol. 136 However, these mutations are less common compared to UVB-triggered CPDs. Molecular repair mechanisms of UV-induced damage include nucleotide excision repair for bulky CPDs and base excision repair for oxidized bases such as 8oGs. Furthermore, failure of DNA repair could result in mutations and become deleterious if it affects key oncogenes or tumor suppressors, such as p53. Clinically, host defense against UVR can be observed through sunburned keratinocytes that undergo apoptosis to avoid conversion into metastatic disease. 136 Melanocytes experience comparatively slower turnover to keratinocytes and are, thus, less likely to experience apoptosis and more likely to develop into its malignant form.
Genetic mutations may not be the only factor that drive UV-induced tumorigenesis. There is evidence that epigenetics, the heritable changes in DNA expression, may be affected by UV irradiation. Epigenetics involve DNA modifications such as methylation of CpG islands along the DNA transcript or chromatin packing through histone acetylation and deacetylation. It has been discovered that following UV treatment, various tumor suppressors are inactivated by DNA methylation, including CDKN2A and PTEN. 72 Further inactivating mutations in histone deacetylases such as HDAC1-3 maintain chromosomes in the relaxed state, increasing its accessibility to transcription factors that promote melanoma development. Other genetic risk factors to melanomagenesis have been determined in UVR models by additional genetic modifications, such as inactivating Ink4a/Arf, CDK4, or CDKN2A, which decreased the latency time to metastasis.21,138
Chemically induced mouse models
Induction of melanoma by UV is the most clinically relevant model to observe melanomagenesis. There are other methods of initiating melanoma, although they are less useful at revealing physiologically accurate tumor behavior. DMBA, an immune-suppressing polycyclic aromatic hydrocarbon, and TPA, a phorbol ester, can both be applied topically to induce skin irritation and black lesions that develop into melanoma. 69 They are most commonly used in combination with UV and other genetically engineered models and are still relevant for studying the effects of immune therapies on tumor growth. A disadvantage to the use of chemicals for melanoma induction is that cells arising from these lesions are nonpigmented, and application of chemical agents is not as homogeneous as UV irradiation. 73
Conclusion
While cell culture models and in vitro studies are valuable and essential for testing potential therapies and understanding the biological processes, molecular physiology, and pathology, as well as effects of gene alterations, it is impossible to fully recapitulate the complexity of the whole organism and the microenvironment in which tumors develop. Thus, there is a growing need for the development of effective and efficient in vivo model systems that share substantial similarities with the human melanomas so that it is as relevant as possible to increase and improve our understanding of the biology of this disease. There are many advantages of using mouse model systems to study melanoma development. In the last decade, numerous models have been developed that not only permit direct testing of new antimelanoma therapies to determine efficiency and toxicity, but also allow control of gene expression or loss.
With the help of several melanoma mouse models, there have been major advances in the diagnosis, treatment, and prevention of melanoma. All the model systems possess unique advantages and disadvantages, necessitating the use of each melanoma model as appropriate. It is still important to be seen if targeted patient therapy can be effectively tailored through the use of a personalized mouse model to maximize treatment quality, ultimately resulting in cures for this currently untreatable disease. The collective knowledge gained from the use of each model described above will ultimately bring us closer to developing more effective treatment modalities for patients with advanced melanoma.
Author Contributions
Wrote the first draft of the manuscript: OFK, FDN, AS. Contributed to the writing of the manuscript: OFK, FDN, MAN, AS. Made critical revisions and approved final version: OFK, FDN, MAN, AS. All authors reviewed and approved of the final manuscript.
Footnotes
Abbreviations
Acknowledgements
We thank Dr. Kent Vrana, Chair, Department of Pharmacology, The Pennsylvania State University College of Medicine, Hershey, PA, USA for providing support for this manuscript.