
Abstract
The detection of supernumerary marker chromosomes (SMCs) in prenatal diagnosis is always a challenge. In this study, we report a paternally inherited case of a small SMC(15) that was identified in prenatal diagnosis due to advanced maternal age. A 39-year-old woman underwent amniocentesis at 16 weeks of gestation. A fetal abnormal karyotype − 47,XX,+mar − with one sSMC was detected in all metaphases. Since this sSMC was critical in the parental decision to continue or interrupt this pregnancy, we proceeded to study the fetus and their parents. Cytogenetic and molecular analyses revealed a fetal karyotype 47,XX,+mar pat.ish idic(15)(ql2)(D15Zl++,SNRPN−-), in which the sSMC(15) was a paternally inherited inverted duplicated chromosome and did not contain the critical region of Prader–Willi/Angelman syndromes. Moreover, fetal uniparental disomy was excluded. Based on this information and normal obstetric ultrasounds, the parents decided to proceed with the pregnancy and a phenotypically normal girl was born at 39 weeks of gestation. In conclusion, the clinical effects of sSMCs need to be investigated, especially when sSMCs are encountered at prenatal diagnosis. Here, although the paternal sSMC(15) was not associated with an abnormal phenotype, its characterization allows more accurate genetic counseling for the family progeny.
Introduction
Small supernumerary marker chromosomes (sSMCs) are defined as extra and abnormal chromosomes whose content cannot be typically determined by conventional chromosome-banding techniques. Generally, their size is approximately smaller or equal to the size of chromosome 20 in the same metaphase spread.1,2 Supernumerary marker chromosomes (SMCs) are found in ∼0.043% of live births and ∼0.075% of prenatal cases and are seven times more prevalent in intellectually disabled patients. 3 They can either be present additionally in an otherwise normal karyotype, a numerically abnormal karyotype (like Turner or Down syndrome), or a structurally abnormal but balanced karyotype with or without the formation of ring chromosome. 4 Approximately 70% of SMCs are de novo and 30% are inherited. 2 The most common SMCs are derived from acrocentric chromosomes and have a satellited or bisatellited structure. Chromosome 15 accounts for the highest percentage (∼50%) of this group.3,5 SMCs derived from chromosome 15 - SMC(15)s - are found in the majority of dicentric cases in which one centromere is inactivated. By conventional cytogenetics, SMC(15)s can be classified into two main groups: small SMC(15)s, which are metacentric chromosomes without euchromatic material and do not contain the Prader–Willi/ Angelman critic region (PWACR), and large SMC(15)s, which are acrocentric chromosomes containing copies of 15q11–q13 region (OMIM #608636). Small SMC(15)s can be familial or de novo and are not directly associated with an abnormal phenotype. In contrast, large SMC(15)s are typically de novo and associated with an abnormal phenotype.6,7
sSMCs are a major challenge in cytogenetic diagnostics and genetic counseling, especially in the prenatal period, because phenotypic abnormalities depend on several factors. Such factors include inheritance, chromosomal origin, content, and structure of the marker. 1 Here, we report a case of a paternally inherited sSMC(15), identified in prenatal diagnosis due to advanced maternal age.
Case Presentation
A 39-year-old pregnant woman (Fig. 1, individual I.1) was carefully monitored due to advanced maternal age, in the obstetric consultation performed at the Hospital of Divino Espírito Santo of Ponta Delgada, the sole existing hospital in the Azorean Island of São Miguel, Portugal. This was the third pregnancy of a nonconsanguineous couple who had two previous healthy children: a 15-year-old female (Fig. 1, individual II.1) and a 12-year-old male (Fig. 1, individual II.2). The investigations were conducted during the prenatal period. The amniocentesis was performed at 16 weeks of gestation (Fig. 1, proband II.3). The fetal karyotype was obtained from an in vitro culture of amniotic fluid cells after 9, 11, and 12 days. Conventional cytogenetics revealed an abnormal karyotype − 47,XX,+mar − with one sSMC detected in all metaphases (Fig. 2A), thereby making the possibility of mosaicism unlikely. In order to determine from which chromosome the sSMC was derived, Nucleolus Organizer Region (NOR) banding and Fluorescence In Situ Hybridization (FISH) analyses were carried out. The results revealed a bisatellited chromosome 15: SMC(15). Molecular investigation by FISH (Fig. 2B) and PCR were also carried out in the fetus to detect whether the critical region of Prader-Willi/Angelman syndromes were present. Together, these analyses showed a fetal karyotype 47,XX,+mar pat.ish idic(15) (q12)(D15Z1++,SNRPN-), in which the small SMC(15) was an inverted duplicated chromosome that did not contain the PWACR.
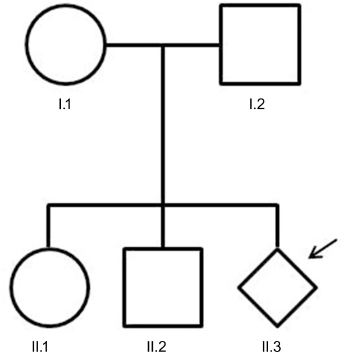
Pedigree of the family with the proband (II.3) indicated by a black arrow.

Fetus (II.3) standard karyotype by G-banding (
Considering that this SMC was critical in the parental decision regarding the continuation or interruption of this pregnancy, both parents were studied by cytogenetics, FISH, and molecular biology. The mother's karyotype was normal (46,XX), but an identical sSMC was found in the father's karyotype [47,XY,+idic(15)(q12)] (Fig. 3A and B). Further, we investigated the uniparental disomy for chromosome 15 in the fetus by comparing the fetal and parental genotypes of seven specific Short Tandem Repeats (STRs). Five of which were located in PWACR. The results showed that three STRs were informative and confirmed biparental contribution to the fetus.

Father's (I.2) standard karyotype by G-banding (
Based on these data – the sSMC(15) without PWACR in fetal and paternal cytogenetic analyses and the exclusion of uniparental disomy for chromosome 15 in the fetus – and on normal obstetric ultrasounds, the parents decided to proceed with the pregnancy. A caesarean section was scheduled at 39 weeks of gestation owing to breech presentation. A phenotypically normal girl (Fig. 1, proband II.3) was born with a birth weight of 2,600 g and an Apgar score of 9/10 at 1 minute and 5 minutes, respectively. A few years later, in reproductive age, both siblings – individuals II.1 and II.2 – decided to be studied on their own will. Cytogenetic analyses showed that the younger brother's karyotype was normal; however, the older sister was found to be a carrier of the same marker [47,XX+idic(15)(ql2)].
Discussion
The identification of sSMCs has improved with the application of modern molecular techniques, such as FISH analysis. Nevertheless, the interpretation of their clinical effects still remains highly problematic, especially when sSMCs are encountered at prenatal diagnosis. 8 sSMCs with low proportion of euchromatin, for example, chromosomes 14 or 15, entail a low risk for phenotypic abnormalities. 9 The large SMC(15)s contain the gene coding for the small nuclear ribonucleoprotein polypeptide N (SNRPN; OMIM *182279) and are tetrasomic for the Prader–Willi syndrome (OMIM #176270) or Angelman syndrome (OMIM #105830) critical region. Based on the parental origin, the larger markers are known to cause a phenotype involving some combination of mental retardation, seizures, autistic features, and growth retardation. 7 There are some exceptional cases of normal phenotypes associated to five or more copies of Prader-Willi region. 10 The small SMC(15)s do not contain SNRPN and are usually associated with a normal phenotype. Although, in some cases, they can be associated with deletions and uniparental disomy of chromosome 15, their frequency in Prader–Willi syndrome cases is greatly increased compared with the normal population (1:40 as opposed to 1:52,000).11,12 The familial case described here could be included in the second group, since no phenotypic abnormalities were found in the three sSMC(15) carriers: proband, her father, and her older sister.
The diagnosis of sSMC during prenatal period is challenging, especially due to its prognosis after birth. Here, we report a case of one sSMC diagnosed in this critical phase, prompting us to investigate several characteristics, such as inheritance, chromosomal origin, content, and structure. Most reports concern de novo or maternally inherited sSMCs. The peculiarity of this clinical case is its paternal origin, which is less frequent. This case supports the literature in two aspects: sSMC(15)s that do not contain PWACR generally have a normal phenotype, 7 and sSMCs transmitted by normal carriers to their offspring are not commonly correlated with clinical problems. 13 In this family, genetic prenatal counseling can be offered to their progeny. Although no phenotypic abnormalities were found, in the literature, it is widely assumed that sSMCs confer a small risk of congenital anomalies above the baseline risk of general population. 14
Conclusion
The majority of small SMCs is derived from chromosome 15 and is usually rare when inherited from the father. In the present case report, we emphasize the difficulty in dealing with a marker chromosome identified during prenatal diagnosis, and we underline the challenge that sSMCs are for genetic counseling. Together, these aspects may offer an additional educational benefit for medical students, physicians, and other healthcare professionals.
Consent
Written, informed consent was obtained from the mother after careful consideration for publication of this case report.
Author Contributions
Conceived and designed the experiments: AP, CA. Analyzed the data and wrote the first draft of the manuscript: BCSM. Contributed to the writing of the manuscript and made critical revisions: LMV. Agreed with manuscript results and conclusions: AS. All the authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
We thank Dr Lúcio Borges and Dr Paula Melo of the Department of Gynecology and Obstetrics, Hospital of Divino Espírito Santo of Ponta Delgada, EPE, Azores Islands, Portugal, for advice on prenatal diagnosis. We also thank students Arya Ibrahim Kermanshah (Pennsylvania State University), Ruben Neves (Fairfield University), Henna Chandel (University of Kentucky), Silvia Vilas Boas (University of Toronto), and Victoria Santos (Dominican University of California) for revising the English language manuscript.