
Abstract
We report a case of sirenomelia sequence observed in an incident of preterm labor during the 29th gestational week. According to some authors, this syndrome should be classified separately from caudal regression syndrome and is likely to be the result of an abnormality taking place during the fourth gestational week, causing developmental abnormalities in the lower extremities, pelvis, genitalia, urinary tract and digestive organs. Despite recent progress in pathology, the etiopathogenesis of sirenomelia is still debated.
Introduction
The sirenomelia sequence, also known as “mermaid syndrome”, is a polymalformative syndrome where the most noticeable (but inconstant) aspect is the fusion (more or less complete) of the lower limbs. It is a rare syndrome (0.8 to 4.2/100,000 births)1,2 which continues to cause many controversies concerning its etiopathogenesis. Its classification as a variant of caudal regression syndrome (CRS) is still debated, despite the conclusions of Stevenson. 3 The same applies regarding its relationship with narrow pelvis syndrome and VATER (vertebral defect, anal atresia, interauricular communication; interventricular communication, tracheal and esophageal atresia, and renal or radial agenesis) syndrome.
We report a case of sirenomelia sequence observed in the obstetrical department of the Aziza Othmana University Hospital of Tunis. Through this case, we will try to shed more light on the classification, etiopathogenesis and management of this syndrome.
Observation
Mrs SK, a woman 25 years old without any noticeable pathological precedent (no diabetes; no teratogenic product use), married for three years to a cousin of the third degree and secondiparous (she had previously given birth to a boy in apparently good health), had been transferred to our department for preterm labor in the seventh month of pregnancy.
The first examination showed a 25 cm uterus, a live fetus and a woman in the first stages of labor. The ultrasound examination performed before the start of tocolysis (LOGIQ 400 Pro 2002 version 5.01 OB/GYN System M-Mode, Color Doppler, PW Doppler, Automatic Tissue Optimization. Reconditioned color Doppler, V5.01, 2 probes connectors, a 3.5 MHz Convex probe for trans-abdominal scanning and a 5 MHz probe for trans-vaginal scanning) showed a single pregnancy at its 29th week of gestation, with an anhydramnios, no bladder and no kidneys, and the ultrasound showed a heterogeneous intra-abdominal image of 4 cm (blind bowel), and a single femur was observed. The absence of amniotic fluid hindered a better morphological study.
Anhydramnios, renal agenesis and the absence of bladder led us to give up the tocolysis and let the labor progress. The delivery occurred three hours after admission. The newborn weighed 1450 g and was polymalformed with a poor Apgar score (cyanosis, nasal flaring, global hypotonia, cardiac pulse >100 and irregular breathing), and died 20 minutes after birth from respiratory distress, probably owing to pulmonary hypoplasia.
The pathological examination of the newborn revealed:
Morphotype abnormalities (Figs. 1 and 2):
Potter's face (ocular hypertelorism [a], low-set ears [b], receding chin [c] and flattening of the nose [d]). Absence of urinary orifice [e]. Anal imperforation [a], two slender limbs [b].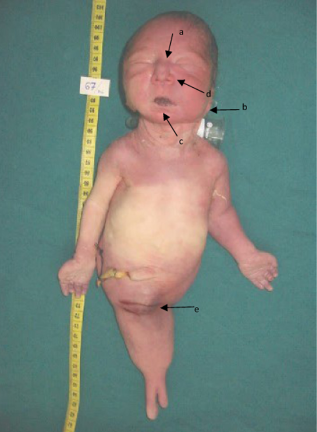

undetermined sex (no external genital organs)
a single inferior limb: an ectopode “mermaid” terminating in two slender limbs (stumps)
a Potter's face (ocular hypertelorism, low-set ears, receding chin and flattening of the nose)
anal imperforation.
absence of a urinary orifice.
Internal abnormalities (Figs. 3 and 4)
Blind bowel [a], normal liver [b], normal heart [c], renal agenesis [d], absence of rectum [e], aorta [f]. Tubular bladder.
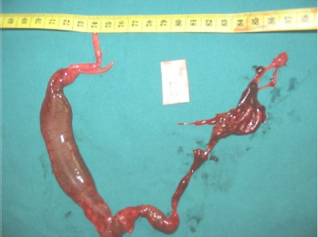
a permeable esophagus
bilateral pulmonary hypoplasia
normal heart and cardiac vessels
normal stomach, liver, biliary ducts and pancreas
two plane-shaped suprarenal glands
an accessory spleen.
differentiated male gonads
absence of a rectum.
a blind bowel forming a hemorrhagic mass of 4×3 cm, containing meconium
renal and ureteral agenesis.
a tubular bladder.
a single umbilical cord artery.
absence of renal arteries.
a single non-branching aorta terminating the right iliac artery.
absence of the inferior mesenteric artery.
The X-ray of the skeleton revealed (Fig. 5):
Skelton radiograph: Sacral agenesis [a], single femur [b], vertebral dysgenesia [c].
chest hypoplasia
a single femur
sacral agenesia
vertebral dysgenesia (L4–L5)
Discussion
Embryology and classification
The abnormalities of embryologic development leading to the sirenomelia sequence occur about the fourth week of gestation, and concern the medio-posterior mesodermic axis and the caudal blastem. 4 At this gestational age, the cesspool (cloaca) is formed, the kidneys are located in the pelvis, while the gonads are intra-abdominal. It seems logical that any damage to the caudal extremity of the embryo would affect the development of the external and internal genital organs (except the gonads, which are intra-abdominal), the terminal bowel, the bladder, the kidneys and the pelvic bones.
Before the publication of Stevenson's studies about sirenomelia's pathological features in 1986, all caudal extremity development anomalies were grouped under the term caudal regression syndrome (CRS) or sirenomelia sequence. This condition is characterized by a spectrum of abnormalities ranging from anal imperforation in its most mild form to full sirenomelia with fusion of the two lower limbs, which constituted the most severe form. 35
Since this publication, some authors such as Jones et al 4 have adopted a new classification which distinguishes the following:
Caudal regression syndrome (CRS) (also called caudal dysplasia sequence). The principal elements of this are defects in the development of the sacrum, up to agenesia, and alterations of the spinal cord with, as a corollary, more or less severe urinary incontinence and misplacement of the lower limbs. Renal agenesia and anal imperforation are inconstant characteristics. CRS is compatible with life, but at the price of heavy motor and sphincterian sequelae. 1
The sirenomelia sequence. This is associated with renal agenesia, an absence of both external and internal genital organs (with differentiated gonads: the sex ratio is 3 boys: 1 girl), 4 anal imperforation with absence of a rectuma dysgenesia of the sacrum to agenesia, sometimes associated with lumbar vertebral dysgenesia and a variable degree of atrophy and an inconstant fusion of the lower limbs (Classification of Stocker and Heifetz: Table 1).6,7 The inconstant fusion of the lower limbs and their misplacement have two possible explanations: an absence of the cleavage of the common bud of the lower limbs by allantois, or an absence of the caudal extremity, which prevents the normal rotation of the lower limbs, thus leading to their fusion on the median line. 6
Classification of sirenomelia by Stocker and Heifetz 6 .

Other authors, such as Stanton et al 1 and Adra et al 8 continue to adopt the old classification, which considers sirenomelia (as described in our case) as the most severe form in the continuum constituted by CRS.
In addition to belonging to CRS, another problem is raised that is the inclusion of sirenomelia within the VATER syndrome because of its frequent association with esophageal atresia and cardiac malformations 4 or its inclusion within the Small Pelvic Outlet syndrome (a narrowing of the pelvis with the ischions getting closer (to fusion), anal imperforation, renal agenesia or dysplasia, urinary tract dysplasia and anomalies of the external genital organs). 9
Etiopathogenesis
Several etiologic factors have been suggested; however none of them explains all the anomalies encountered in the sirenomelia sequence:
Maternal diabetes is the only maternal disease known to be associated with sirenomelia (2% of cases), although this is associated more frequently with CRS (22% of cases).5,10 The hypothetical role of insulin has been removed, since maternal insulin does not pass through the placental barrier and fetal insulin secretion begins only from the 10th gestational week.
Teratogens: retinoic acid, cadmium and cyclophosphamide have been implicated in the genesis of sirenomelia in mice and hamsters.11–13 However, no case of sirenomelia in humans has been observed after incidental maternal exposure to these products. Cocaine, organic solvents of fat and appetite suppressors (diethylpropion) have been implicated in some cases of CRS in humans.14,15
Genetic factors: several family cases of CRS have been reported, as well as some cases of minor vertebral abnormalities in the parents, suggesting a possible genetic transmission. Several modalities of transmission have been suggested: dominant sex-linked transmission, multifactor polygenetic transmission, and dominant autosomic transmission with a variable expression and an attenuated penetrance. Welch et al 16 suggested an association between maternal diabetes and a genetic predisposition.
The vascular steal theory: this theory was initially suggested in 1927 by Kampmeier and was called “nutritional deficit”. It was reintroduced in 1986 by Stevenson 3 under the term “vascular steal”. Stevenson relied on observations taken from the autopsies of 11 cases of sirenomelia, which determined that the blood returns to the placenta through a large vessel deriving from the vitelline artery and starting from the abdominal aorta (just under the diaphragm). The abdominal aorta does not give any collateral. A mega-artery ensures the function of both umbilical arteries and diverts the blood flow on the embryo's caudal extremity toward the placenta, causing a nutritional deficit and a lack of development of the caudal extremity. Sirenomelia is quite often accompanied by a unique umbilical artery. 6 Our observation of a single umbilical artery, renal agenesis and the absence of renal arteries might support this theory, but the presence of a tubular bladder cannot be explained by vascular steal. The gonads are vascularized, in principle, by collateral arteries of the abdominal aorta starting from the top of the renal arteries. The preservation of the gonads supposes the preservation of their vascularization. The observations of Stevenson reveal the absence of collaterals coming from the abdominal aorta; such observations explain the preservation of gonads (as in our case) only poorly, thus throwing a doubt on the vascular steal theory. The vascular steal theory also fails to explain the frequent association of sirenomelia with other abnormalities such as cranial, cardiac and esophageal defects. 5
Adra et al consider Stevenson's theory of the vascular steal as a possible etiopathogenic of CRS, while Stanton maintains that this theory cannot explain all the abnormalities encountered in the sirenomelia sequence and that the observations of Stevenson are a consequence and not a cause of the syndrome.1,8
The pressure theory (not widely accepted): this implicates certain amniotic forces which act on the caudal extremity of the embryo leading to its hypoplasia. Gardner et al 17 suggest that an excessive distention of the neural tube at its caudal extremity leads to lateral rotation of the mesoderm, thus causing a fusion of the lower limbs and a closure of the primitive bowel and the urethra.
Twin pregnancies and sirenomelia sequence: 9% to 15% of the cases of sirenomelia are associated with twin monozygotic pregnancies. The relative risk is multiplied 100-fold in the case of twin pregnancies. 18 A genetic mechanism seems less probable because of the absence of family cases.
Prognostic
Sirenomelia is fatal in most cases because of the characteristic pulmonary hypoplasia and renal agenesia. About 50% of the children are born alive after eight or nine months of pregnancy. Death occurs in the five days following birth. 6 Post-natal management requires the presence of kidneys, even if they are dysgenesic. 1 Murphy et al 19 reported one case where a child born with sirenomelia survived; the infant was neurologically normal and had fused lower extremities, an imperforate anus, colon atresia, bilateral fused pelvic kidneys with renal dysplasia and sacral dysplasia, and genital abnormalities. Laparotomy and colostomy were performed, and an eventual separation of the lower extremities was planned. Clarke et al 20 report on a three-month-old infant whose sirenomelia was diagnosed prenatally The infant was neurologically normal and has fusion of the lower limbs with associated renal dysplasia, an imperforate anus, pelvic and sacral dysplasia, and genital abnormalities. The infant's anomalies are compatible with life and surgical separation of the lower limbs was planned. Managing sirenomelia is difficult and quite costly, requiring several interventions, the results of which are unpredictable (in the case described by Stanton, 1 the patient had five interventions between the age of 15 days and 4 years; she continues to be bedridden and dependent).
Antenatal diagnosis (Table 2)
Prenatal ultra sonographic diagnosis of sirenomelia.

Since the prognosis is bad, the management of sirenomelia is difficult, with unpredictable results. It seems more than justified to put the emphasis on antenatal diagnosis in order to choose the cases with better prognosis. Antenatal diagnosis is based on a morphologic ultrasound study (oligohydramnios, bilateral renal agenesis, a single lower limb, a unique umbilical artery, absence of a bladder, undetermined external genitalia, anorectal atresia, lumbosacral agenesis). In sirenomelic fetuses, bilateral renal agenesis causes severe oligohydramnios, hindering a reliable sonographic evaluation of the lower extremity in the second and third trimester. In some cases, bilateral renal agenesis is the only sonographic finding. Amnioinfusion and high-frequency transvaginal ultrasonographic probes have proven to be very useful in such a situation.5,10,21 Whereas oligohydramnios is a marker of renal agenesis or non-functioning kidneys in the second half of pregnancy, in earlier gestational stages, other contributors to the production of amniotic fluid production are present. Therefore, early in the second trimester, the amount of amniotic fluid should be sufficient to allow detection of sirenomelic abnormalities. Early diagnosis will allow termination of the pregnancy at an early stage, with minor risks and discomfort for the patient. 5
The MRI (magnetic resonance imagery) permits the evaluation of visceral lesions, avoiding the call for amnioinfusion (which carries a risk of fetal injury, infection, membrane rupture and placental injury).
It seems justifiable to insist on the screening of sirenomelia in diabetic women as well as the screening of diabetes in pregnant women.
The therapeutic decision depends on the gestational term at the time of diagnosis, and on the severity of the malformative syndrome and the parents' wish. Genetic counseling should be proposed because of the reoccurrence risks (estimated to be 3%–5%).
Conclusion
This rare syndrome, the sirenomelia sequence, will continue to divide the scientific community as to its etiopathogeny and its classification. However, to be pragmatic, it seems that the emphasis should be placed both on prenatal diagnosis and genetic to ensure an optimal management that would consequently be less demanding both from a psychological point of view and a health cost point of view.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material. Written consent was obtained from the patients parents to reproduce information and photographs appearing in this work.