
Abstract
Chronic neutrophilic leukemia (CNL) is a rare myeloproliferative neoplasm (MPN) that represents a diagnostic dilemma for both clinicians and pathologists. Because this disease entity is very rare, and because its diagnosis is by exclusion, it is important for clinical hematologists and hemato-pathologists to be familiar with CNL when approaching patients with MPNs and persistent neutrophilia. A woman in her 40s who was incidentally found to have leukocytosis was referred to the hematology service at the National Center for Cancer Care and Research for evaluation. Complete blood count revealed hyperleukocytosis with predominant neutrophilia. Peripheral blood and flow cytometry did not show any evidence of lymphoproliferative disorder or myeloblasts. Bone marrow aspirate and biopsy revealed a hypercellular marrow with myeloid hyperplasia. Cytogenetics revealed normal karyotype. Tests for both Janus kinase mutation JAK2 V617F and rearrangement of the genes BCR–ABL1, platelet-derived growth factor receptor-α (PDGFRα), PDGFRβ, and fibroblast growth factor receptor-1 (FGFR1) were negative. Thereafter, the diagnosis of CNL was reached. She was treated with pegylated interferon alpha-2a, with very good hematological response. To the best of our knowledge, this is the first case of CNL reported among the Arab population.
Background
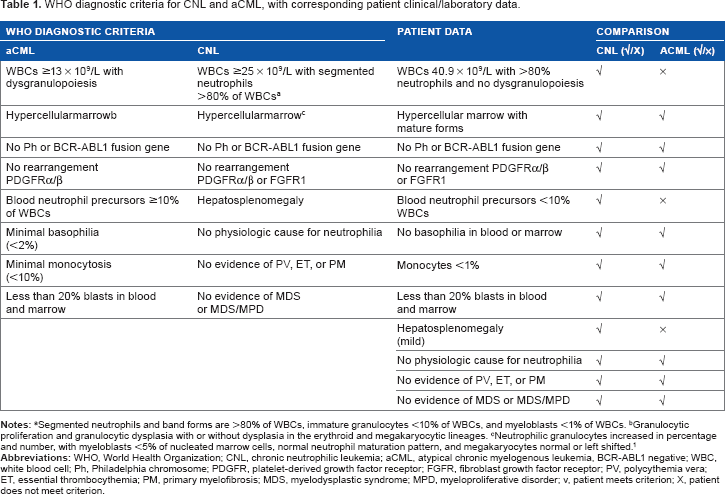
Chronic neutrophilic leukemia (CNL) is a rare myeloproliferative neoplasm (MPN). CNL diagnosis is only reached after excluding reactive neutrophilia, MPN, myelodysplastic syndrome (MDS), or overlap of MDS/MPN. Absence of BCR-ABL1, platelet-derived growth factor receptor-α (PDGFRα), PDGFRβ, and fibroblast growth factor receptor-1 (FGFR1) rearrangements is also one of the minimal diagnostic requirements for CNL. 1 According to the World Health Organization (WHO), as of 2008, the diagnostic criteria for CNL are the following: leukocytosis >25 × 109/L; >80% segmented neutrophils; and <10% immature granulocytes, in the absence of granulocytic dysplasia, myelodysplastic changes in other myeloid lineages, monocytosis, eosinophilia, or basophilia. 1 Additional clinicopathologic characteristics of CNL include splenomegaly, elevated vitamin B12 level, and neutrophilic leukocytosis that is characterized by toxic granulation and Döhle bodies.
Case Presentation
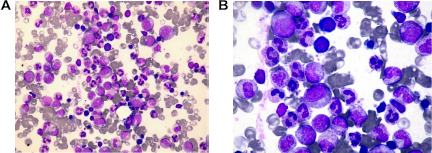
A woman in her 40s was incidentally found to have leukocytosis. She was referred to the Hematology service at the National Center for Cancer Care and Research for evaluation. Her clinical examination was unremarkable and there was no hepatosplenomegaly. Most notable among the first set of studies was an abnormal white blood cell (WBC) count of 40.9 × 103/μL (reference range: 4.0- to 11.0 × 103/μL). The differential count revealed 95% bands/segmented neutrophils, 4% lymphocytes, and 1% monocytes, eosinophils, and basophils. Hemoglobin (Hb) level was 10.1 g/dL and platelet count was normal. Her peripheral blood smear revealed neutrophilic leukocytosis with increased toxic granulation. Neutrophil precursors were <1%, with occasional myelocytes noted on scanning. No circulating myeloblasts or neutrophil dysplasia was noted. The bone marrow aspirate was hypercellular with myeloid hyperplasia, with a predominance of mature neutrophils and no relative increase in blast count (blasts = 1%). Toxic granulations were seen in neutrophils (Fig. 1A and B). The myeloid: erythroid ratio was 7.5: 1. The erythroid series was sparsely represented but did not show any morphologic abnormalities. The majority of megakaryocytes were normal in size and morphology, with only minor hypolobulation on a subset of cells (Fig. 2A and B). No increase in eosinophils, basophils, plasma cells, or mast cells was observed. Sea blue histiocytes were not seen. Stainable iron was markedly reduced without any ringed sideroblasts. Significant dysplasia was not present in any of the cell lineages. The bone marrow core biopsy was hypercellular for age, with a cellularity estimated at 75%–85% with neutrophilic proliferation and adequate megakaryocytes (Fig. 3A). There was no increase in myeloblasts, eosinophils, basophils, or mast cells. Only minimal focal reticulin fibrosis was noted in some regions. Immunohistochemical staining performed on the core biopsy showed predominance of myeloperoxidase-positive myeloid cells, without any increase in cluster of differentiation-34 (CD34)-positive cells (Fig. 3B). The conventional marrow karyotype was 46, XX, with no abnormalities noted. A t(9;22) translocation was not identified by either polymerase chain reaction or fluorescence in-situ hybridization methods. Mutation analyses for Janus kinase-2 (JAK2) and PDGFRα/PDGFRβ were similarly negative. The mutation analysis for the colony-stimulating factor-3 receptor gene (CSF3R) was performed by bidirectional sequencing method. The mutation hot spots exon 14 and exon 17 of this gene were analyzed. This assay has a typical sensitivity of 10%–15% for detecting mutated CSF3R DNA. CSF3R was studied and the result was negative; similarly, FGFR1 was investigated and the result was negative. Computerized scans of the chest, abdomen, and pelvis were negative for lymphadenopathy or hepatosplenomegaly. Positron emission tomography– computed tomography (PET/CT) scans were negative. Blood, urine, stool, and sputum cultures were done repeatedly, as well as sputum cultures for acid-fast bacilli, Mycobacterium tuberculosis, and Brucella, with sustained negative results. The diagnosis of CNL was thereafter reached. The patient was treated with pegylated interferon alpha-2a (Pegasys®), as per Yassin et al. 2 This therapy comprised the following protocol 2 : 50 μg once weekly for 2 weeks, then 135 μg once weekly for 6 weeks, and finally 135 μg every 2 weeks.
(

(

(
Our patient showed hematological remission in terms of normalization of WBCs because her WBC count remained below 11,000; her platelets were normal and remained so all through the treatment and her Hb level remained >10 g/dL, with no symptoms or infections and with excellent clinical condition. The patient was offered a repeat bone marrow test but she was reluctant. As per our knowledge, this is the first case report with interferon alpha-2a; what was reported previously by Meyer et al. 3 was therapy using interferon alpha 2b.
Discussion
Myeloproliferative disorders comprise a range of conditions, ie, BCR-ABL-positive chronic myelogenous leukemia (CML), CNL, polycythemia vera, primary myelofibrosis, essential thromobocythemia, chronic eosinophilic leukemia not otherwise specified, mastocytosis, and unclassifiable MPN. 4 In the WHO classification of myeloid disorders, CNL is recognized as an MPN characterized by sustained neutrophilic leukocytosis, hepatosplenomegaly, and bone marrow granulocytic hyperplasia without evidence of dysplasia, BCR-ABL1, or rearrangements of PDGFRα, PDGFRβ, or FGFR1. This diagnosis is dependent on the exclusion of underlying causes of reactive neutrophilia, particularly if evidence of myeloid clonality is lacking. The lack of a specific molecular marker has left the diagnosis to be largely one of exclusion. Recently, the molecular landscape shifted with the discovery of specific oncogenic mutations in the CSF3R in CNL patients. 5 Being a diagnosis of exclusion, CNL identification is difficult for both clinician and pathologist. Our patient presented with leukocytosis. In clinical practice, neutrophilia most commonly relates to leukemoid reactions due to chronic infections, inflammatory diseases, or various types of malignancies. 6
In our patient, there were no symptoms or signs of inflammations, and PET/CT scanning was performed to rule out hidden malignancies, the result of which was negative. Clini-copathologic characteristics of CML include splenomegaly and a neutrophilic leukocytosis with left shift, and these were ruled out by negative BCR-ABL, absence of Philadelphia chromosome, and normal cytogenetic analysis. Negative JAK2 V617F helps to exclude other myeloproliferative neoplasms such as polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Myeloid neoplasm with PDGFRα and PDGFRβ were ruled out by the negative results for molecular markers. CNL is a rare MPN, with only ∼200 patients reported to date, mostly from case reports and small case series. 1 Thus, 50%–60% of patients with CNL or aCML harbor mutations in the receptor for CSF3R (GCSFR). Under normal circumstances, the CSF3R ligand, granulocyte-colony-stimulating factor (G-CSF), promotes growth and survival of myeloid precursor cells, ultimately leading to differentiation of these myeloid precursors into neutrophils. Deletion of CSF3R leads to neutropenia in mouse models. 7 In addition to regulating normal neutrophil homeostasis, G-CSF levels rapidly increase during infection, resulting in elevated levels of neutrophils as a component of the immune response. 8 The normal role of CSF3R in promoting neutrophil production is biologically consistent with our observation of CSF3R activating mutations in hematologic malignancies characterized by high levels of neutrophils. Our patient was tested for this mutation and found to be negative. The absence of hepatosplenomegaly is not against CNL. Persistence of neutrophilia for more than 1 year and absence of all secondary causes make CNL the most likely diagnosis because its diagnosis is only by exclusion. Additional factors of CNL usually present with splenomegaly but absence of splenomegaly, normal cytogenetics, and molecular markers that rule out CNL are not seen.
WHO diagnostic criteria for CNL and aCML, with corresponding patient clinical/laboratory data.
No standard of care exists for CNL or aCML. Therapy has primarily consisted of cytoreduction by hydroxyurea or other oral chemotherapeutics, as well as use of interferon–α.9–11 These agents can elicit improvement in blood counts but exhibit no proven disease-modifying benefit. Although splenic irradiation and splenectomy may provide transient palliation of symptomatic splenomegaly, the latter has been associated with anecdotal worsening of neutrophilic leukocytosis in CNL. The limited experience with induction-type chemotherapy for blastic transformation is generally poor, with death related to resistant disease or regimen-related toxicities. Allogeneic transplantation may result in favorable long-term outcomes in selected patients, particularly when undertaken in the chronic phase of disease. 9 Our patient, who was recently married few months before diagnosis, required different treatment options. These options were explained to her, and she opted for pegylated interferon alpha-2a. This therapy was started as per Yassin et al. 2 The treatment was well tolerated by the patient and she successfully achieved good hematological response.
In summary, even in the era of molecular testing, in the case of this woman in her 40s, the diagnosis of CNL represents a diagnostic difficulty. In addition, the treatment of CNL remains experimental, with no standard of care due to the nature of the disease and its rarity.
Author Contributions
Conceived and designed the experiments: MAY. Analyzed the data: MAY. Wrote the first draft of the manuscript: MAY, SK. Contributed to the writing of the manuscript: SK, AY, AM, AN, AAL, AAB, ATS. Agree with manuscript results and conclusions: MAY, SK, AAB, ATS, ND, AAL, AM, AN, AY. Jointly developed the structure and arguments for the paper: MAY, SK. Made critical revisions and approved final version: MAY, ATS. All authors reviewed and approved of the final manuscript.