
Abstract
Here we present the case of a 60-year-old woman with a rare sellar region atypical teratoid/rhabdoid tumor (AT/RT), complicated by lung metastasis and treated with neurosurgery, radiotherapy, and chemotherapy. The patient had recurrent headache associated with left cavernous sinus syndrome after a previous endonasal transsphenoidal resection for a presumptive pituitary macroadenoma. Pituitary magnetic resonance imaging showed a tumor regrowth in the original location with a haemorrhagic component involving the left cavernous sinus. A near complete transsphenoidal resection of the sellar mass was performed followed by 3 months of stereotactic radiotherapy. Because of a worsening of the general clinical conditions, respiratory failure, and asthenia, the patient underwent a contrast enhanced computer tomography of the whole body which showed the presence of lung metastasis. The histopathological diagnosis on samples from pituitary and lung tissues was AT/RT. The patient survived 30 months after diagnosis regardless chemotherapy. In the adult, the AT/RT should be considered as a possible rare, aggressive, and malignant neoplasm localized in the sella turcica.
Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) occurs mainly in in children younger than 3 years of age. This tumor is similar in appearance to a rhabdomyo-sarcoma but the cells differ from the expected morphological and immunohistochemical features of muscle. A tumor composed of rhabdoid cells in the central nervous system (CNS), was first reported in 1985. The name atypical teratoid/rhabdoid tumor (AT/RT) exemplifies the tumors disparate mixtures of rhabdoid, primitive neuroepithelial, mesenchymal, and epithelial components, although the tumors may be composed entirely or partly of rhabdoid cells.1–4 The histologic and immunohistochemical features of this neoplasm have been extensively described.5–10 The polymorphous histology prompts misdiagnosis of AT/RT as primitive neuroectodermal tumor (PNET), medulloblastoma, glioblastoma, choroid plexus carcinoma, or malignant teratoma5,6,11–13 The development of AT/RTs have been associated with a specific genetic alteration, that is, mutations of the INI1/hSNF5 gene located on chromosome 22q11.2. 14 The INI1/hSNF5 gene encodes a component of the SWI/SNF chromatin remodelling complex, interacting with sequence specific DNA binding proteins such as c-Myc and EBNA-2. 15 This genetic hallmark of AT/RT, either mutation or deletion of both copies of the INI1/hSNF5 gene, is seen in approximately 70% of tumors. A smaller number have reduced expression at the RNA or protein level, indicative of a loss-of-function event. Loss of INI1 protein expression in the tumor cells is seen in all cases of AT/RT. Demonstrating this by immunohistochemistry has been suggested as a useful marker to distinguish AT/RTs from other malignant CNS tumors. 16 Loss of expression of INI1 as detected by immunohistochemical staining correlates with deletion and mutations of the INI1 gene. 17
Multiple therapeutic approaches have been attempted over the last 2 decades in an attempt to increase survival in these patients without much success. 18 Radiation therapy seems to be the most important component of therapy. There is no accepted standard chemotherapy, but intensive high-dose chemotherapy may be more effective. Efforts to delay radiation often fail, and most reported cases of survivors, even when including high-dose chemotherapy, frequently require repeat surgery and radiation. Overall, patients usually succumb to their disease between 6 months and 1 year from diagnosis.
Clinical Case
We describe the clinical history of an obese 60-year-old woman, married with 3 children, who previously underwent an endonasal transsphenoidal resection for an intrasellar nonfunctioning presumptive pituitary macroadenoma invading the left cavernous sinus. At the first clinical observation, the patient presented with frontotemporal stabbing pain, insomnia, left diplopia, and convergent strabismus. After neurosurgery, the pathology study showed that the mass could be classified as atypical pituitary adenoma ACTH-, GH-, PRL-, TSH-, FSH-, chromogranin A-, with light cytoplasmatic positivity for pan-cytokeratin, LCA-, Vimentin+, S100-, EMA-, CD34-, CD1a-, p53+(10%), ki67+(30%).
One month after resection, the patient was readmitted to our clinic for headache associated with pain unresponsive to indomethacin. The patient showed left eye lachrymation, photophobia, nausea, diplopia, and signs of paresis of the IV pair of cranial nerve. She underwent magnetic resonance imaging (MRI) of the sella turcica with 1.5T superconductive units before and after an intravenous injection of contrast medium (0.1 mmol/kg of body weight). MRI showed the tumor regrowth in the original sellar location with a haemorrhagic component involving the left cavernous sinus and encasing the internal carotid artery (Fig. 1). A new transsphenoidal resection was performed with a near complete resection of the sellar mass. The pathology examination identified a neoplastic fibrous stroma tissue infiltrated by a population of pleomorphic cells with abundant eosinophilic cytoplasm and hyperchromatic nuclei concluding as presumptive atypical pituitary adenoma. After 2 months, she again presented headache, abnormal right-directional gazing, diplopia in all directions of gaze, and exophthalmos. The MRI showed a large and dishomogeneus sellar mass with lateral, inferior, and anterior extension involving the cavernous sinus, the sphenoidal bone with sinus invasion, the left orbital apex with orbital fissures, and the optic canal (Fig. 2). The patient underwent 3 months of stereotactic radiotherapy (17 Gy to the periphery and 34 Gy to the center of the residual tumor). A few months later, because of a worsening of the general clinical situation, dyspnea, and asthenia, a contrast enhanced computed tomography (CECT) scan of the whole body revealed several metastases involving the pulmonary parenchyma bilaterally (Fig. 3). The patient underwent a thoracoscopic atypical lung resection. All the surgical specimens including the sellar and lung tissue neoplasm were reviewed by a referent neuropathological centre. The final diagnosis was secondary lung localization of AT/RT with primary pituitary localization. Therefore, systemic antineo-plastic therapy was started with doxorubicin (DOXO) and vinorelbine (VNL) (DOXO 37.5 mg/mq gg 1–2; VNL 30 mg/mq gg 1–8). After 9 cycles, the total body CECT control demonstrated a mild progression of the sellar mass and stability of the lung metastasis. Stereotactic radiotherapy (SRT) was then performed on the residual sellar mass. As a complication of SRT, the patient showed cerebrospinal fluid fistulas treated surgically. After treatment, the patient underwent an additional CECT, which showed a progression of the disease. A CT of the head revealed a huge invasive sellar and extra sellar mass that eroded the sphenoid bone, with invasion of the ethmoid and erosion of the anterior cranial fossa. A large component of the mass occupied the nasopharinx (Fig. 4); a progression of the lung localization was also observed. A therapy consisting in carboplatin AUC 5/taxolo 175 mg/mq every 21 days was started. During the second cycle, the clinical condition of the patient deteriorated. She presented with hyposthenia and paraesthesia of the upper and lower limbs, hearing impairment, reduction of visual acuity, nose bleeding, and dyspnea. Unfortunately, 30 months after the first neurosurgical treatment, she died.

Coronal CE SE T1W (
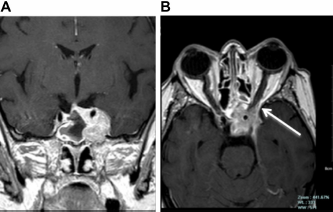
Coronal (
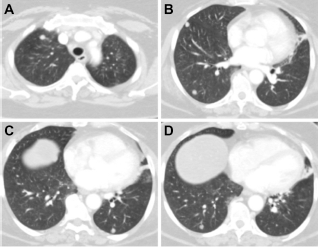
Pulmonary CECT showing several nodular metastases in axial slices of both lungs.

CECT of the head showing a huge invasive sellar and nasopharinx mass that eroded the sphenoid bone, including clivus, sellar floor, and ethmoid with erosion of the anterior cranial fossa.
Laboratory Studies
The biochemical evaluation before neurosurgery revealed a normal adenohypophyseal function. After the second neurosurgery resection, a condition of global adeno-neuro-hypophyseal hypofunction (ACTH < 1 pg/mL [reference range 5–60 pg/mL], FT3 0.97 pg/ mL [reference range 1.8–4.6 pg/mL], FT4 0.78 ng/dL [reference range 0.93–1.71 ng/dL], FSH 1.4 mUI/mL [reference range 1.5–12.4 mUI/mL], LH 0.1 mUI/mL [reference range 1.7–8.6 mUI/mL], cortisol 0.2 μg/dL [reference range 7–25 μg/dL], urinary free cortisol < 20 μg/dL [reference range 30–130 μg/dL]) 19 was found and a replacement therapy was established. Molecular studies performed on tissue obtained by lung biopsies demonstrated a deletion of chromosome 22q (probe Vysis LSI 22q11.2/Tel Vysion 22q13:ratio 22q11/22q13 = 0.69). 20
Pathological Findings
The primitive diagnosis of the sellar lesion after the first neurosurgery was nonfunctioning presumptive atypical pituitary adenoma, with immune-histochemical negativity for GH, PRL, LH, FSH, TSH, and ACTH (data not shown). The recurrent sellar lesion (Fig. 5A and B) and multiple lung metastases (Fig. 5C–E) were then reviewed at our institution and showed a neoplasm composed of small-to medium-sized polygonal cells arranged in cords or ribboned pattern with oval nuclei, vesicular chromatin, and eosinophilic cytoplasm. In some fields, large cells with frank rhabdoid appearance were evident and showed eccentrically placed vescicular nuclei containing prominent nucleoli, with paranu-clear inclusion and eosinophilic cytoplasm. These cells grew singly in a fibrous stroma or in clusters. There were atypical mitosis and apoptotic figures. The proliferation labelling index, measured by MIB-1 monoclonal antibody to the Ki67, was 25%. Immunohistochemical evaluation showed focal positivity for epithelial membrane antigen, cytokeratin, smooth muscular actin, and gliofibrillary acidic protein. These multidifferentiation features in accord with the histological findings suggested the possibility of an AT/RT, which was validated by the lack of immunostaining for INI1 protein in neoplastic cells compared with positivity of the internal control represented by the endothelial cells (Fig. 5B and E). The final histological diagnosis was a World Health Organization (WHO) grade IV AT/RT with primary location in the sella turcica and secondary location in the lung. Written consent was obtained from the husband of the patient to reproduce information and photographs appearing in this work.
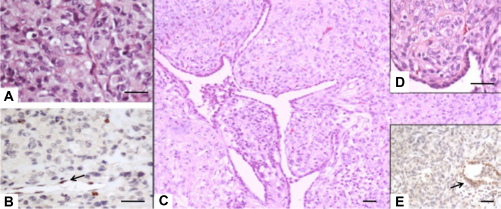
Hematoxylin and eosin staining (
Discussion
Here we describe the case of a middle-aged obese woman presenting with a sella turcica AT/RT with suprasellar extension. AT/RT is a rare, aggressive, and malignant disease typical of infancy and childhood affecting the central nervous system, first defined as “atypical teratoid tumor.” 21 Based on histological evidence of diffuse proliferation of atypical large cells with eccentrically located nuclei, prominent nucleoli, and abundant cytoplasm (rhabdoid cells), the disease was later referred to as AT/RT of the brain in order to emphasize the variable combination of rhabdoid cells, epithelial cells, and primitive neuroectodermal, as well as mesenchymal features, 1 Its occurrence in adult patients is extremely rare, as 95% of reported patients are 5 years old or younger.8,22 Only 32 documented cases have been described in adults to date, usually located in the cerebral hemisphere, cerebellum, and spinal cord, even if unusual locations such as the mediastinum, liver, neck, shoulder, and retroperitoneum have been described.4,23 A unique presentation of a rare case of adolescent AT/RT confined to the leptomeninges with no obvious intraparenchymal primary lesion and no focal neurological deficit has been also reported. 24 Occurrence of AT/RT in the sellar region is rare, with only 7 cases reported so far.25,26 The rare incidence of this lesion, in particular in adults, may lead to its misdiagnosis as in the case reported here where the early diagnosis was pituitary macroadenoma with suprasellar extension. However, our patient's age and the sellar origin of her tumor may argue against a diagnosis of AT/RT and dictate a differential diagnosis with pituitary carcinoma or metastasis. The definitive diagnosis might be carried out through loss of immunohistochemical INI1 protein expression due to mutations of the INI1/hSNF5 gene.27,28 The prognosis of AT/RT is poor both in adults and in the pediatric population, although long-term survival is possible in adult AT/RT cases after a combined approach including surgery, adjuvant radiotherapy, and chemotherapy.25,29 Postoperative radiotherapy is crucial in the treatment of AT/RT,30,31 and our patient underwent this treatment to have the remaining tissue removed closer to the sinus cavernous not excised by the neurosurgeon. This combined approach, performed before the final pathological diagnosis, allowed us to observe a better prognosis in the patient with a survival of 30 months. In conclusion, AT/RT should be considered in the differential diagnosis of aggressive tumors with sella turcica localization even in middle-age individuals.
Author Contributions
Conceived and designed the experiments: CM, FG, LG. Analyzed the data: DL, FS. Wrote the first draft of the manuscript: LC, PDG. Contributed to the writing of the manuscript: MC, MF, RM. Agree with manuscript results and conclusions: SU, MA. Jointly developed the structure and arguments for the paper: CM, SU, LG. Made critical revisions and approved final version: CM, SU, FG, LG. All authors reviewed and approved of the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.