
Abstract
The updated classification of idiopathic interstitial pneumonias (IIPs) in 2013 by American Thoracic Society/European Respiratory Society included several important revisions to the categories described in the 2002 classification. In the updated classification, lymphoid interstitial pneumonia (LIP) was moved from major to rare IIPs, pleuroparenchymal fibroelastosis (PPFE) was newly included in the rare IIPs, acute fibrinous and organizing pneumonia (AFOP) and interstitial pneumonias with a bronchiolocentric distribution are recognized as rare histologic patterns, and unclassifiable IIP (UCIP) was classified as an IIP. However, recent reports indicate the areas of concern that may require further evaluation. Here, we describe the histopathologic features of the updated IIPs and their rare histologic patterns and also point out some of the issues to be considered in this context.
Introduction
In 2013, an updated classification of idiopathic interstitial pneumonias (IIPs) 1 was provided as a supplement to the previous 2002 IIP classification. 2 The revision preserved the basic contents of the 2002 classification but also included several modifications. Here, we describe the histopathology of major IIPs, with the entities newly included in rare IIPs and unclassifiable interstitial pneumonia (IP). 1
Summary of the Updated Classification of IIPs Published in 2013
In the 2013 updated classification, although most histologic patterns described in the 2002 classification were still listed as major IIPs, lymphoid interstitial pneumonia (LIP) was recategorized among the rare rather than the major IIPs (Table 1). In addition, a newly recognized condition, pleuroparenchymal fibroelastosis (PPFE), was included in the rare IIP category. Unclassifiable IP was already included in the 2002 classification but referred only to truly unsolvable conditions. In the updated classification, unclassifiable IP is one of three major categories of IIPs (Table 2). Another significant change was the grouping of major IIPs. Six major IIPs were divided into three major groups. Thus, idiopathic pulmonary fibrosis (IPF) and nonspecific interstitial pneumonia (NSIP), respiratory bronchiolitis–interstitial lung disease (RB-ILD) and desquamative interstitial pneumonia (DIP), and cryptogenic organizing pneumonia (COP) and acute interstitial pneumonia (AIP) were entered into the three groups of chronic fibrosing IIPs, smoking-related IIPs, and acute/subacute IIPs, respectively. Acute fibrinous and organizing pneumonia (AFOP) and IPs with a bronchiolocentric distribution were newly included as rare histologic patterns.
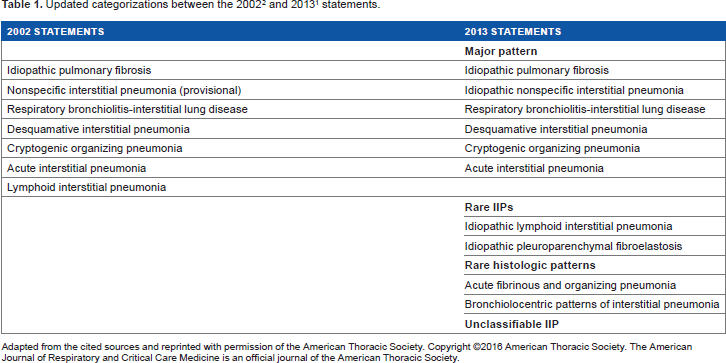
Adapted from the cited sources and reprinted with permission of the American Thoracic Society. Copyright ©2016 American Thoracic Society. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
Categorization of major IIPs (from the 2013 statement). 1

Adapted from the cited sources and reprinted with permission of the American Thoracic Society. Copyright ©2016 American Thoracic Society. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
Chronic Fibrosing IIPs
Idiopathic Pulmonary Fibrosis/Usual Pneumonia Pattern
IPF is the most important and common form of chronic interstitial lung disease (ILD). As described in the 2002 statement, the pathology of IPF is that of usual interstitial pneumonia (UIP). 2 The term “UIP” was originally introduced by Liebow and Carrington, 3 who defined it as “chronic lung fibrosis of the common or usual type.” This definition covers a broad category of chronic ILDs. In the 2011 IPF guidelines, 4 the diagnosis of IPF was modified to include either radiographic or pathologic UIP. Therefore, in cases of radiologic UIP, a multidisciplinary discussion does not require pathologic confirmation of UIP. This modification may be a crucial one, as some cases of radiologic UIP may include conditions with other etiologies, such as subclinical hypersensitivity pneumonia or interstitial pneumonia with autoimmune features (IPAF). 5 In rare cases, hypersensitivity pneumonia or IPAF are identified solely based on the pathologic findings. Furthermore, minor modifications may be needed for the current diagnostic algorithm indicated in the IPF guidelines.
Histopathologic Features of UIP
The key histopathologic feature of UIP is its heterogeneous appearance, as seen at low magnification. The areas of scarred fibrosis are immediately adjacent to normal-looking parenchyma. The fibrotic areas are mainly located in the peripheral zones of secondary lobules along with perivenular areas sandwiched by primary lobules (Figs. 1A and 1B). These areas are bordered by various levels of tiny subacute injured foci. These fibroblastic foci (FF) are composed of fibroblasts and myofibroblasts and are covered by type II pneumocytes or squamous metaplastic epithelia (Fig. 1C). They occur more frequently and more widely in UIP than in fibrotic NSIP and correlate with impaired pulmonary function, such as vital capacity and the carbon monoxide diffusion capacity of the lungs.6,7 Based on several reports, it was shown to be a poor prognostic factor of UIP.7,8 FF is thought to be the core pathogenic event in this disease.9,10 The dense collagen and FF may be accompanied by microscopic honeycomb changes. The latter, in which the aggregates of cystic spaces are surrounded by scarred fibrosis, is the end-stage appearance of fibrosis. Honeycomb cysts are often covered by columnar ciliated epithelium (Fig. 1D). Their mucous or proteinaceous fluid filling can make them difficult to identify on high-resolution computed tomography (CT). The distinction between traction bronchiectasis and honeycomb cysts may be an important one, but it is often difficult to recognize it. The pathologic findings of honeycomb cysts and traction bronchiolectasia may differ from the CT findings. Elastic staining (eg, elastica van Gieson stain) is useful for the diagnosis of UIP, as it highlights the distribution of fibrosis and the nature of the architectural destruction by showing thick fragmented elastic fibers (Fig. 1E). In the majority of UIP cases, elastic fibers accumulate in peripheral areas of the secondary lobules (Fig. 1F); this may be the result of structural collapse and is a predictor of poor prognosis. 11 The progression of UIP is shown schematically in Figure 2.
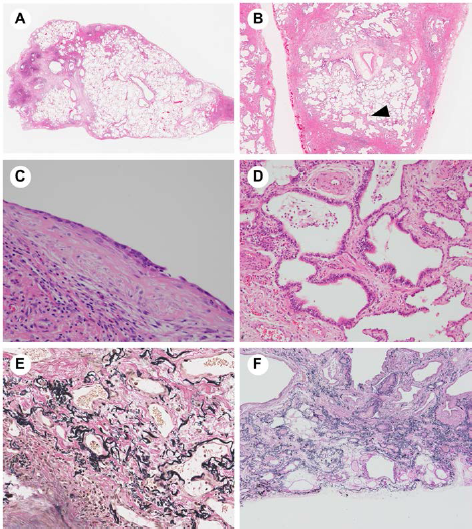
Pathology of usual interstitial pneumonia (UIP): (

Pathologic schema of UIP. In the early phase, patchy dense fibrosis involves peripheral and perivenular areas, but the architectural destruction is mild. Here, a fibroblastic focus is located between the fibrotic area and normal lung. In the later phase, structural remodeling due to wider fibrosis causes shrinkage, where that honeycomb cysts develop in association with traction bronchiolectasia. Red lines: arteries; blue lines: veins; yellow lines: lymphatic ducts; and green areas: FF.
Histologic Criteria for UIP Pattern in the 2011 IPF Guidelines
One of the accomplishments of the 2011 guidelines was the definitions of “definite,” “probable,” “possible,” and “not” UIP, based on the level of diagnostic confidence. The histopathologic criteria for UIP pattern (Table 3) are mostly identical to those in the 2002 guidelines. Important new additions are the criteria to rule out UIP. These include hyaline membrane, organizing pneumonia (OP), granulomas, marked inflammation distinct from honeycombing, predominant airway-centered changes, and other features suggestive of an alternative diagnosis. In the presence of any of these findings, the correct diagnosis is “not UIP.” Yet, despite the delineation provided by these criteria, the diagnosis of “not UIP” is ultimately a subjective one.
Histopathologic criteria for UIP pattern (from the 2011 IPF guidelines). 4

Adapted from the cited sources and reprinted with permission of the American Thoracic Society. Copyright ©2016 American Thoracic Society. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
Sampling Methods for the Diagnosis of UIP
The pathologic diagnosis of UIP is based on the findings obtained from either surgically resected or autopsied specimens. Transbronchial lung biopsy (TBLB) or needle biopsy samples are usually inadequate for the diagnosis of UIP because they are mainly derived from the center of the secondary lobules and usually contain insufficient tissue to determine whether the histologic criteria are fulfilled, as these were established based on peripheral regions of the lung lobules. A recent technique that enables a diagnosis of ILD, including UIP, is cryobiopsy. The specimens obtained with this technique are larger than those obtained using TBLB, and the tissue architecture is preserved without crush artifacts.12–14
Nonspecific Interstitial Pneumonia
There are two major types of NSIP: (1) cellular NSIP, characterized by diffuse inflammatory cell infiltration, and (2) fibrosing NSIP, in which there is dense fibrosis.15–17 Often, there is a mixture of both, but these cases are also categorized as fibrosing NSIP. According to the current statement, NSIP is included in the category of chronic fibrosing IP. Therefore, the pure form of cellular IP may have to be excluded from the diagnosis.
The fibrosis in NSIP is uniformly and diffusely spread throughout the lung and pulmonary lobules (Fig. 3A). 15 The basic architecture of the lung is relatively maintained, and there is less destruction of the normal structure than that in UIP. The alveolar septa are thickened by an inflammatory cell infiltrate or fibrosis, in which the precise structure of the alveolar sac may disappear due to collapse. This condition is referred to as “simplification” of the lung (Fig. 3B). The distinction between UIP and fibrosing NSIP is important for pathologists.15,16 The key to discriminating between the two conditions is the accurate recognition of the diffuse nature of NSIP. 18 Thus, NSIP has a uniform appearance, with temporal homogeneity along with the lack of “completely normal” lung. Smooth muscle hyperplasia, FF, and aggregation of elastic fibers, which are the features of UIP, are not observed in most cases of NSIP. Nonetheless, the distinction between UIP and NSIP can be difficult, especially in advanced disease, as honeycomb-like cystic changes with distortion of the lung architecture may be present in both. A schematic view of the progression of NSIP is shown in Figure 4. NSIP can also coexist with other histologic patterns, such as OP, UIP, and diffuse alveolar damage (DAD). 19

Pathology of the nonspecific interstitial pneumonia (NSIP) pattern: (

Pathologic schema of the NSIP pattern. In the early phase, fibrosis involves the alveolar septa uniformly and diffusely. OP foci (green opacities) may be seen in the airspaces. In the later phase, cystic changes due to traction, with mild distortion of the lung architecture, may occur. In the end-stage NSIP, honeycomb changes will eventually develop.
Smoking-Related IIPs
Respiratory Bronchiolitis-Interstitial Lung Disease
The histology of this mostly smoking-associated condition is centrilobular accentuated abnormalities.20,21 Alveolar macrophages with dusty brown pigment and black particles in and around respiratory bronchioles are the characteristic and most significant finding. Mild lymphocytic infiltration and/or minimal fibrosis of the respiratory bronchioles and adjacent alveolar septa may be found but are not mandatory for a diagnosis of RB-ILD (Figs. 5A and 5B). Macrophage features are mostly identical to those seen in DIP; in both conditions, the cells will show slight positivity for iron staining. 20 In fact, the differences between RB-ILD and DIP are not remarkable, and some pathologists include the two conditions with a spectrum, considering its separation based on local histologic differences. In general, the alveolar macrophages and fibrotic changes are more limited to peribronchiolar areas in RB-ILD than those in DIP.20,21 The association with emphysema is fairly common in both diseases.

Pathology of respiratory bronchiolitis-interstitial lung disease (RB-ILD): (
Desquamative Interstitial Pneumonia
Histologically, DIP lesions show uniform and diffusely distributed abnormalities (Fig. 6A). The lone distinctive biopsy finding is extensive airspace filled by pigmented alveolar macrophages throughout the sample. The term “desquamative” is, in fact, a misnomer. It derives from the frequent association of DIP with hyperplastic type II cell proliferations, in which the airspace-filling cells were initially thought to be desquamative type II cells before immunohistochemical staining convincingly showed that they are macrophages. There is a strong morphologic resemblance between alveolar macrophages and the hyperplastic type II cells of DIP. 22 Mild IP along with the presence of lymphoid follicles and moderate-to-severe emphysema in the background alveolar septa are common findings (Fig. 6B).18,23 Structural renovation of the basic lung architecture is mild. A uniform pattern of fibrosis is often seen in DIP with hyalinized thickening of the alveolar septa that spread from subpleural areas to central zones of the lung. Completely normal alveolar septa are not usually present; however, when evident, they will be abruptly separated by interlobular septa. In the long-term follow-up, the development of cystic spaces with dense fibrosis, mimicking honeycomb changes, may be seen in DIP. 2 As mentioned earlier, the distinction between DIP and fibrotic NSIP associated with a DIP reaction may be difficult, and interobserver agreement regarding these two conditions is typically low.
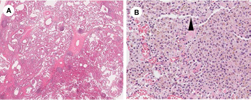
Pathology of desquamative interstitial pneumonia (DIP): (
Acute/Subacute IIPs
Cryptogenic Organizing Pneumonia
OP consists of an airspace-filling fibroblastic proliferation along with various degrees of edema and inflammatory cell infiltration. It refers to the histologic reaction and can be seen in any type of inflammatory or repair process, including localized infection or infarction. OP is also a common reaction in various types of IP. The term “COP” should therefore be carefully and strictly used only for those cases of multifocal IIPs. COP has a patchy distribution and involves several lobules; its border around normal lung areas is relatively clear (Fig. 7A). Even inside the affected area, the lung architecture is mostly maintained (Fig. 7B). In prototypical OP, the polypoid spherical organization is composed of myofibroblasts and small number of lymphocytes in an edematous matrix (Figs. 7C and 7D). The surface of the OP is usually covered by type II cells. 24 Sometimes, there will be mural incorporations of the airspace organization. In this case, OP may mimic a fibroblastic focus of UIP, where such confirmation of the absence of dense collagen depositions immediately next to the OP foci is important. Another important consideration in the differential diagnosis of OP is the organizing phase of DAD. If the abnormal OP-related change is seen throughout the submitted slides, the diagnosis may be organizing DAD.
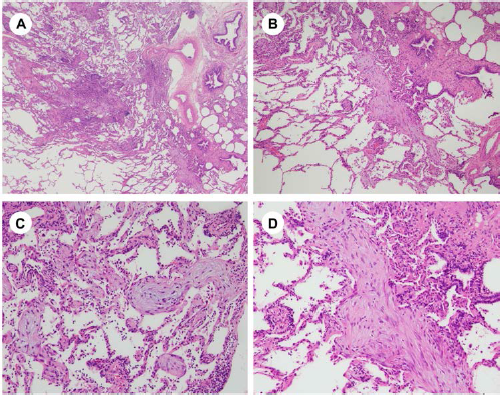
Pathology of cryptogenic organizing pneumonia (COP): (
Acute Interstitial Pneumonia
The prototype pathology of AIP is uniformly distributed DAD. Histologically, depending on the time elapsed from disease onset, AIP is divided into the exudative, organizing (or proliferative), and fibrotic phases. In the exudative phase, the basic lung structure is maintained. Common changes are stromal edema, epithelial necrosis and denudation, collapse of the alveolar sacs which results in dilatation of the alveolar ducts, hyaline membrane formation mostly in the alveolar ducts, and effusions into the alveolar space (Fig. 8A). However, DAD often has other causes. In these cases, there is more remarkable hyaline membrane formation and inflammatory cell infiltration. The organizing (proliferative) phase is characterized by a more pronounced alveolar collapse, the proliferation of fibroblasts within alveolar septa, hyperplastic type II pneumocytes, and vascular endothelial cell swelling with/without a fibrin thrombus (Fig. 8B). Squamous metaplasia may also be observed. The proliferating fibroblasts may form a prominent OP, which can lead to the erroneous pathologic diagnosis of OP. In the fibrotic phase, dense fibrosis is accompanied by microscopic honeycomb changes, which are mostly due to the collapsed alveoli and dilated alveolar ducts. Prominent squamous metaplasia covering the area of microscopic honeycombing is a characteristic feature. Because the distinction between fibrotic DAD and UIP may be difficult, a multidisciplinary discussion with experienced pulmonologists, chest radiologists, and pulmonary pathologists is mandatory.

Pathology of acute interstitial pneumonia (AIP): (
Acute Exacerbations of UIP
The pathology of an acute exacerbation of UIP largely consists of the mixed features of DAD and UIP. DAD is usually seen in nonfibrotic lesions.
In cases marked by repeating episodes of exacerbation, a mixture of the different phases of DAD in the same biopsy may be observed.25,26 Hyaline membranes may be unclear; instead, pathology may show OP or faint changes of acute lung injury, such as interstitial edema or focal airspace fibrin.26,27 The fibrotic areas of UIP may not be present, depending on the biopsy site. A correlation between clinical history and radiologic changes is critical for the accurate diagnosis of an acute exacerbation of UIP.
Rare IIPs
Idiopathic LIP
LIP is generally associated with other clinical conditions, such as collagen vascular disease and malignant lymphoma. As a rare condition, idiopathic LIP has newly been categorized within the rare IIPs. In LIP, there is a marked stromal infiltration by lymphoid cells, with significant infiltration of the alveolar septa (Fig. 9A).1,2 The distribution of the disease is uniform. The main infiltrating cells are lymphocytes, without atypia, plasma cells, and histiocytes. Eosinophils and neutrophils are rare. Occasionally, alveolar septa with a severe lymphoid infiltration may develop into a dense fibrosis. Large lymphoid follicles with germinal centers are frequently present. Infiltration into the pleura or interlobular septa is inconspicuous, but in such cases, lymphoma is the more likely diagnosis. Cholesterol clefts, foreign body reaction, and sarcoid-like granuloma may be sparsely distributed. In the airspace, eosinophilic proteinaceous fluid, well highlighted by immunoglobulin staining, is a common finding (Fig. 9B), as it is a cyst formation without significant fibrosis (Fig. 9C). Around the cyst, there is often a fragmented alveolar sac, as seen in the emphysematous lung. Cases in which there are marked lymphoid follicles along the lymphatic route but without infiltration of the alveolar septa should be categorized as “diffuse lymphoid hyperplasia” and must be separated from LIP.

Pathology of lymphoid interstitial pneumonia (LIP): (
LIP is rarely idiopathic; rather, it is usually a component of a lymphoproliferative disorder, such as lymphoma, multicentric Castleman's disease, or IgG4-related disease, or a pulmonary manifestation of connective tissue disease, such as Sjögren's disease. 28
Idiopathic Pleuroparenchymal Fibroelastosis (iPPFE)
On histology, iPPFE characteristically shows dense elastic fibrosis expanding from the pleura to the lung parenchyma, predominantly in the upper areas of the lung (Fig. 10A). Elastic fiber staining highlights the basic alveolar structure, which is well preserved; the elastic fibers of alveolar septa are uniformly increased, and the airspaces inside the lesion are completely filled with various levels of collagenous fibers (Figs. 10B and 10C).29–31 Thus, iPPFE is a completely different disease from UIP, in which the alveolar architecture is mostly destroyed. iPPFE lesions contain double the amount of elastic fibers seen in UIP. 32 The pleura often, but not always, shows chronic fibrosing pleuritis. The inflammatory cell infiltrate is often faint and is limited to occasional lymphoid aggregations without germinal centers. Traction bronchiectasis is frequent but lacks the surrounding inflammatory changes. The histologic changes observed in iPPFE are mostly identical to those of apical cap, and the purely histologic distinction between the two is extremely difficult. iPPFE cases are mostly limited to upper lung fields, but in some cases, there is an association with different interstitial abnormalities that simulate conventional UIP or NSIP in the lung base. In such cases, whether the patient has iPPFE alone or two different lung diseases is not certain. 31

Pathology of pleuroparenchymal fibroelastosis (PPFE): (
Rare Histologic Patterns
Acute Fibrinous and Organizing Pneumonia
AFOP was first reported as a histologic pattern of acute lung injury and a possible variant of DAD. 33 In AFOP, there is an extensive airspace fibrin exudate that forms “fibrin balls” rather than the organizing fibrosis seen in OP (Fig. 11). Airspace involvement in AFOP is roughly 50%. AFOP differs from DAD by the lack of hyaline membranes and from eosinophilic pneumonia by the lack of eosinophils. 33 The pathologic changes in AFOP are distinctive, but whether it is a distinct IIP is currently uncertain. To date, there has been no convincing report describing the characteristics of AFOP. Its associations with hypersensitivity pneumonia in which acute exacerbation developed, 34 connective tissue disease, 35 and drug reaction 36 have been reported. Infections including pneumocystis may show a similar histology and should be excluded.

Pathology of acute fibrinous and organizing pneumonia (AFOP): (
IPs with Bronchiolocentric Distribution
Bronchiolocentric fibroinflammatory changes, also called as bronchocentric IP, airway-centered interstitial fibrosis, and peribronchiolar–metaplasia-associated ILD have been described in a small number of cases based on retrospective observations.37–39 Follow-up reports are limited, and the clinicopathologic significance of the condition is still uncertain. The data suggest a poor prognosis for patients in whom fibrosis expands to the lung parenchyma. Some cases of IP with bronchiolocentric distribution may be related to specific inhalations, such as tobacco smoking, or agents that give rise to chronic hypersensitivity pneumonia.
Unclassifiable IIP
Cases eventually diagnosed as unclassifiable IIP (UCIP) may include those with inadequate clinical, radiologic, or pathologic data and those in which there is a major discordance between the clinical, radiologic, and pathologic findings. The latter discordance may be due to therapeutic effects, an unusual variant of a recognized entity that is not adequately characterized by the current American Thoracic Society/European Respiratory Society (ATS/ERS) classification, or the presence of multiple radiologic and/or pathologic patterns in one patient with idiopathic disease. According to the ATS/ERS, cases with autoimmune features, such as lung-dominant connective tissue disease, are now classified as IPAF. 5 Whether these cases should be treated as a subgroup of connective tissue disease or IIPs is not clear. There are no established criteria to separate such cases because of a shortage of concrete evidence. Some IPAF cases may also fulfill the criteria of UCIP. 40 Further prospective studies are needed to determine the optimal approach to UCIP, but this confusing category would benefit from a reorganization.
Conclusion
We have described the updated criteria for IIPs and rare histologic patterns, with an emphasis on the histopathologic aspects. The recently updated reports of these diseases have also revealed areas requiring further investigation. We need further studies to improve application of the new classifications.
Author Contributions
Wrote the first draft of the manuscript: MH. Made critical revisions and approved the final version: JF. Both authors reviewed and approved the final version of the final manuscript.
Footnotes
Acknowledgments
We thank all of our colleagues of the Department of Pathology, Nagasaki University, who kindly gave their time and assistance in writing this article.