
Abstract

Introduction
In 2009, chronic wounds had an incidence rate of 6.5 million cases annually in the USA, and $25 billion was expended on their treatment. 1 Audits of community healthcare clinics in the UK in 2005–2007 showed wound care costs ranging from £2.5 to 3.1 million per 100,000 patients or 2%–3% of local healthcare budget expenditure. 2 Unfortunately, almost half of these wounds are nonresponsive to current treatment strategies and they represent a challenging clinical problem. 3 Early and late complications are frequent causes of morbidity and mortality in the clinical treatment of wounds.4,5 Chronic wounds have an incidence rate of 120 per 100,000 people aged between 45 and 65 years, and by the age of 75 years, this rises to 800 per 100,000.5,6 Population dynamics calculated by The World Bank indicates that this latter age group is steadily increasing on a global scale. 7 The cost of treating chronic wounds is escalating due to such aging trends and increasing rates of diseases such as diabetes and obesity, which prolong the treatment of chronic wounds. 1 There is a clear need to better understand the underlying pathobiology of chronic wounds in order to formulate more effective means of treating these conditions. 8 Without such basic studies and their application in the clinic, no meaningful progress will be made in the treatment of wounds.
Definition of wound healing
Mason and Dunhill
9
proposed that the healing response should be considered as a replacement or regeneration of human cells, tissue, or organs to restore normal tissue function.
10
The use of the term
Polymeric HA inhibits platelet degranulation and downregulates inflammation and excessive collagen deposition, which contribute to scarless healing. In contrast, in adult wound healing, chondroitin sulfate (CS) is the major GAG, and platelet degranulation, inflammation, and collagen deposition at the wound site are essential components of the adult wound healing process but contribute to scar tissue formation.
GAGs and wound healing
Four classes of GAGs have been identified, including HA, CS, keratan sulfate (KS), and heparin sulfate (HS). These are composed of characteristic repeat disaccharides, with specific monosaccharides sulfated at C-2, -3, -4, or -6 (Fig. 1). All of these GAGs are widely distributed in connective tissues. An additional carbohydrate recognition motif human natural killer-1 (HNK-1) has a relatively restricted distribution mainly in the peripheral nervous system (PNS)/central nervous system (CNS). HNK-1 has a widespread distribution in neural tissues and is a component of proteoglycans (PGs), cell adhesion molecules, bioactive lipids, structural glycoproteins, and key synaptic enzymes (Fig. 2).
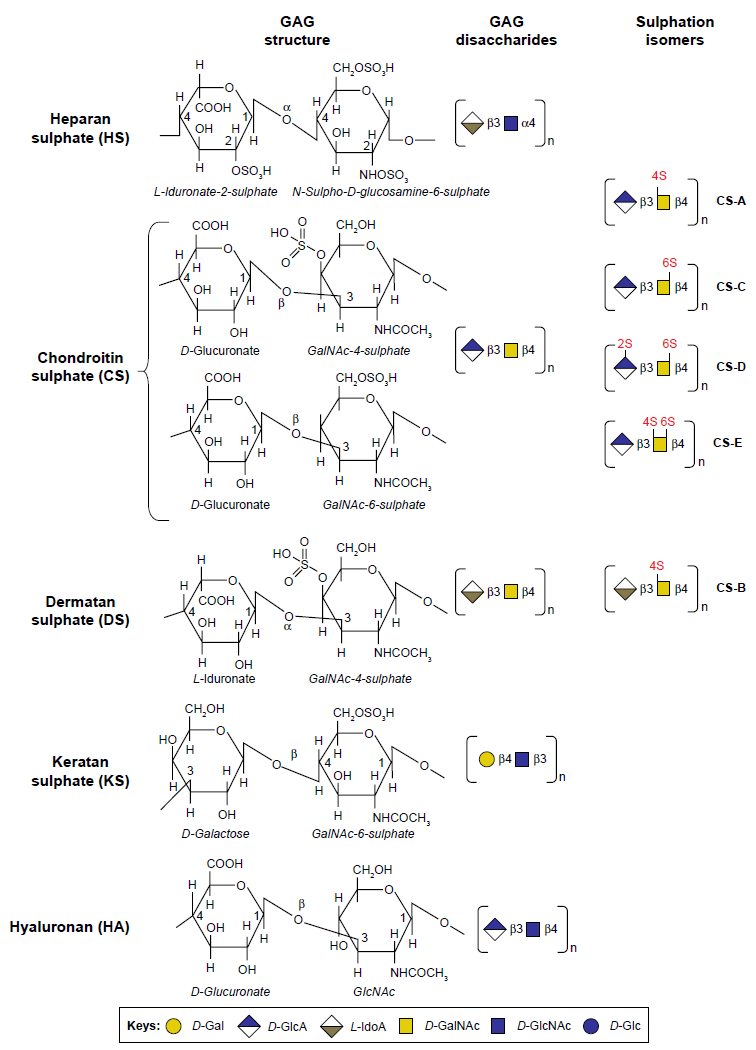
Structures of glycosaminoglycans.
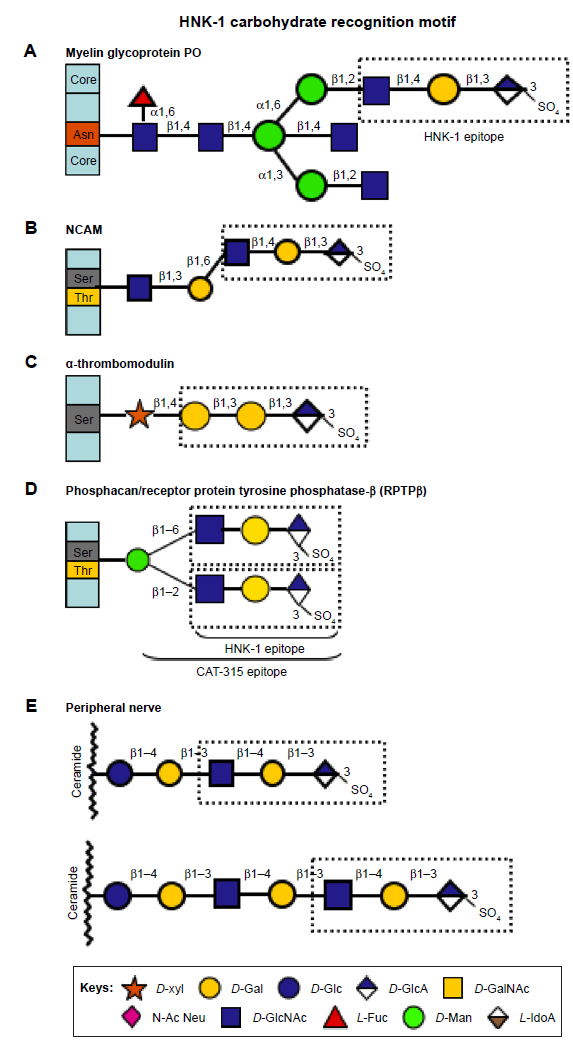
GAG organization in selected neural biomolecules.
The phases of wound healing
Wound healing is a dynamic process with some complexity where devitalized and degraded tissue is replaced with new functional tissue. GAGs have roles to play in all phases of wound healing.
Phase 1.
The initial stage of wound repair lasts one to two hours with formation of a transitional fibrin matrix through activation of the coagulation cascade, cleavage of fibrinogen by thrombin, and formation of a fibrin clot (Fig. 3). HA synergizes with thrombin and promotes clot formation. Soluble fibronectin is cross-linked into the clot, and platelets, immune cells, and mast cells are attracted into this tissue. Clot formation is countered by enzymes of the fibrinolytic cascade, which ensure that excessive thrombus formation does not occlude blood flow. PDGF and TGF-β released from the platelet α-granules and other mediators released at the wound site such as epidermal growth factor (EGF) and IGF-I act as chemoattractants for neutrophils, smooth muscle cells, and fibroblasts and induce cellular proliferation, differentiation, and matrix synthesis. Serpins ensure that excessive fibrinolytic activity does not occur. GAGs have specific roles to play in the regulation of serpins (Table 1). Heparin and HS promote the inhibitory capability of many of the serpins. Antithrombin (AT) (SER-PINC1), heparin cofactor II (HCII) (SERPIND1), plasminogen activator inhibitor-1 (PAI-1), protein C inhibitor (PCI) (SERPINA5), and protease nexin-1 (PN-1) interact with heparin and HS, leading to conformational rearrangements, which improve inhibitory performance. The antithrombotic activity of AT stems from its ability to inactivate thrombin, Factor Xa, and Factor IXa (Fig. 3). In the presence of heparin, the rate of inhibition of thrombin by AT is increased 2000- to 4000 fold, the rate of inhibition of Factor Xa is increased 500- to 1000-fold, and the rate of inhibition of Factor IXa is increased 1 million fold. Kallistatin, a kallikrein inhibitory protein of endothelial cells and SMCs, and α1-proteinase inhibitor also interact with GAGs. Proteases (chymase, tryptase, leukocyte elastase, and cathepsin G) are complexed with granule PGs (serglycin) in mast cells and leukocytes, and degranulation releases these active proteases, which degrade the extracellular matrix (ECM), and inactivates microbial infection at the wound site. Macrophages attracted into the wound site mop up the cellular and ECM debris.
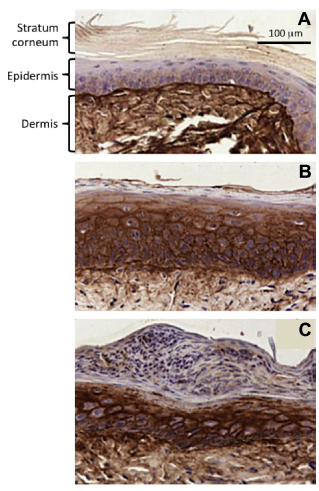
Accumulation of HA in cutaneous wounds. Induction of HA synthesis in the wound site and adjacent areas, which serve as a source of keratinocytes that migrate into the wound site to speed up the re-epithelialization. (A) Normal mouse skin distant to the wound site. (B) Skin adjacent to the wound. (C) The wound site three days after wounding. Normal mouse epidermis has very little HA, but it increases adjacent to the wound site and is associated with epidermal thickening (increased cellular proliferation). Dermal tissue is always rich in HA. Figure supplied courtesy of Professor R.H. Tammi and Professor M. Tammi, Department of Biomedicine and Anatomy, University of Eastern Finland, Kuopio, Finland. 262
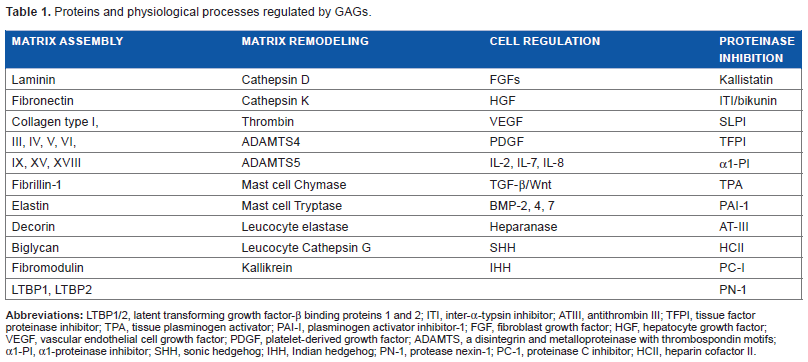
Proteins and physiological processes regulated by GAGs.
Phase 2.
The next phase of wound repair is an inflammatory phase acting over the next 24 hours involving members of the kallikrein protease family, which release kinins at the wound site and other vasodilatory compounds (histamine, prostaglandins, and leukotrienes), increasing vascular permeability. Leukocytes (neutrophils and monocytes), mast cells, neutrophils, and monocytes gain access to the wound site. Neutrophils release IL-1 and TNF-α, inducing inflammation. Neutrophil collagenase and elastase remove damaged tissue from the provisional matrix of the wound site. Mast cells also degranulate at the wound site releasing chymase and tryptase. Monocytes transform into macrophages and phagocytose fragments of denatured ECM debriding the wound site and inactivating any source of microbial infection. They also secrete TGF-β, PDGF, FGF-2, IL-1, and TNF-α, which modulate collagen deposition by fibroblasts and penetration of new blood vessels into the wound site. T-lymphocyte migration into the wound site delivers lymphokines, which modulate fibroblast and endothelial cell activity; fibroblast-activating factor is a stimulatory factor, while IFN-γ is a negative regulator.
Phase 3.
The laying down of new matrix by fibroblasts over the next two to three days restores tissue at the wound site. Matrikines and matricryptins generated at the wound site act as chemoattractants, while other matricryptins act as angiogenesis inhibitors or stimulate collagen synthesis. Fibroblasts, endothelial cells, and keratinocytes produce IGF-I, FGF-2, TGF-β, PDGF, and vascular endothelial cell growth factor (VEGF), promoting cellular migration, proliferation, matrix synthesis, and angiogenesis. A number of PGs are laid down in the wound repair site, and their GAG side chains have roles in the sequestration, stabilization, and activation of these growth factors. The PGs act as scaffolding material in the wound site providing architectural form and stability and a matrix for cellular attachment. Some PGs (hyalectans) form ternary complexes with HA hydrating the tissue promoting cell survival and migration into the repair site.
Phase 4.
The final stage in wound repair is a maturational remodeling stage where recovery of normal tissue form and function occurs. This may be over a protracted period of time up to one year after the initial wounding. New collagen fibrils laid down in the repair zone undergo cross-linking to stabilize collagen fiber bundles and MMPs remodel this repair tissue.
The role of HA in wound healing
HA is abundant in cutaneous wounds and promotes the migration of keratinocytes into the wound site to undertake reepithelialization (Fig. 4). HA (>30 kDa) is a component of the fibrin clot in wound sites synergizing with thrombin during fibrin clot formation, decreasing the lag-phase for clot formation, and increasing the rate and extent of clot formation. The fibrin clot is a transient matrix, which provides hemostasis and attracts platelets, a source of many anabolic growth factors (including TGF-β), which supports wound healing, and chemoattractants, which attract cells into the clot to promote repair. In matrix assembly and cell-mediated wound repair, the GAGs decorate the core proteins of a number of matrix- and cell-associated PGs, which have critical roles in coordinating the replenishment of tissue at the wound site (Table 2). Free GAGs are also present in wounds undergoing repair and are actively secreted by fibroblasts in the wound site. Kosir et al 11 showed that 79.7% of the total sulfated GAGs was present in a free form and 15.6% attached to a PG core protein and CS constituted 79.1% and HS constituted 28.7% of the total free sulfated GAG. Matrix- (perlecan, decorin, and biglycan) and cell-associated PGs (syndecan-2) were decorated with CS and HS. Some of the cell surface PGs are shed from the cell surface as small peptide–GAG complexes, which diffuse to a more remote area in the wound site. Heparanase-1 and -2 release HS and their complexed growth factors in remodeling wounds. Osteoblasts secrete high levels of heparanase-1 and -2 at the bone-cartilage interface. Hypertrophic chondrocytes in this region have abundant levels of pericellular perlecan, and the release of HS chains by heparanase-1 and -2 promotes endothelial cell migration, angiogenesis, and bone formation. HA oligosaccharides (6–10 mer) stimulate scratch wound repair and excisional wound closure by fibroblasts through RHAMM, CD44, and Toll receptor-mediated cell signaling,12–14 and accumulation of M1 and M2 macrophages and M2 TGF-β1 at the wound site promotes this process, 15 whereas 40 mer HA oligosaccharide inhibits wound closure, increases macrophage numbers at the wound site but does not increase TGF-β1 content in the wound or tissue fibrosis. 16 High molecular weight is anti-inflammatory, whereas low molecular weight (<50 kDa) is proinflammatory. Low molecular weight HA is proangiogenic and synergizes with VEGF to promote wound repair. 17 High molecular weight HA promotes the healing of diabetic foot ulcers18,19 through the action of IL-10 20 with adult fibroblasts, a potential mean of improving scarless wound healing 20 HA inhibits fetal platelet aggregation and the release of TGF-β at wound sites contributing to scarless healing 21 and formation of the fibrin clot, which provides hemostasis during the early stages of wound repair (Fig. 4).
Proteoglycans involved in hard and soft tissue repair.
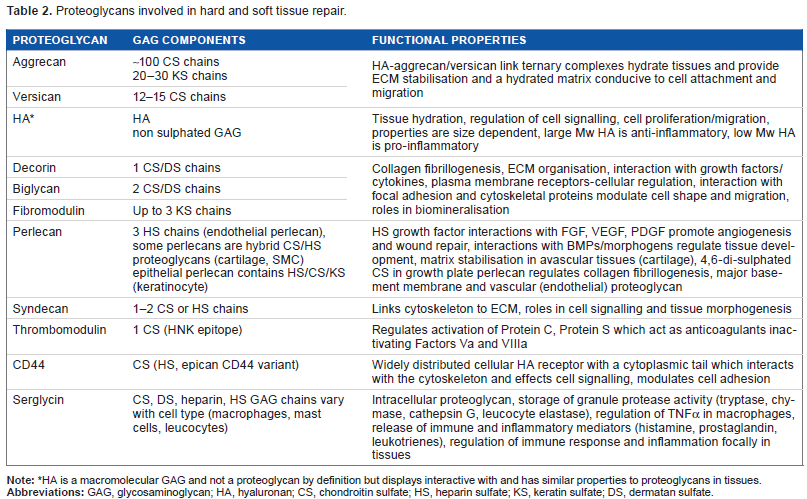
HA is a macromolecular GAG and not a proteoglycan by definition but displays interactive with and has similar properties to proteoglycans in tissues.

Diagram of fracture repair stages (A). Hemotoma formation in fracture site (day 0), cartilage soft callus formation and penetration of new blood vessels (day 7), resorption of soft callus and replacement by bony callus (day 14), and bone remodeling (day 21). Fracture repair in AJ mouse, Safronin-O aniline blue femur cross-section (day 14) (B). Higher power view of (B) depicting enlarged chondrocytes surrounded by a cartilaginous matrix (Safronin-O stain) (C) and femur cross-section (day 21) (D). CT reconstruction of a rat fracture model, cross-sections (day 14) (E and F), and longitudinal sections (day 14) (G). 3D reconstruction of a rat femur (day 21) illustrating the cartilage callus laid down in a femoral fracture site (day 21) (H). Figure modified from Ref. 263
GAG complexity and biodiversity and their roles in wound healing
Four classes of GAGs, including CS, KS, dermatan sulfate (DS), and heparan sulfate (HS)/heparin, have been identified (Fig. 1). A further GAG, HA, occurs devoid of a core protein and is synthesized at the cell surface and extruded out of the cell into the ECM. All other GAG members are synthesized by a complex series of biosyn-thetic steps in the endoplasmic reticulum with 20+ enzymes involved in HS biosynthesis, while CS requires ~10 biosyn-thetic enzymes. GAGs have diverse regulatory roles in ECM assembly, proteinase regulation, cell regulation through the action of growth factors, cytokines, and transcription factors, and proteinase inhibition in the coagulation/fibrinolytic systems relevant to wound repair (Table 1).
GAGs display a considerable degree of structural diversity and complexity encoding a sophisticated information system, which directs cellular behavior. Potentially, 1008 different pentasaccharide sequences are possible in CS/DS and 2916 pentasaccharide sequences are possible in HS. 22 GAGs can explore a varied number of interactive spatial orientations. Their structural form has persisted virtually unchanged throughout vertebrate and invertebrate evolutions. 23 GAGs have essential life-preserving roles to play in a diverse range of physiological processes involving chemokines, morphogens, and growth factors. GAGs are composed of characteristic repeating disaccharides, linear sequences of five to six monosaccharides, typically provide interactive properties for a specific ligand. GAGs are assembled from a limited number of monosaccharides and amino sugars into characteristic disaccharide repeat units (Figs. 1 and 2). Modifications such as N-deacetylation, N-sulfation, epimerization of glucuronic acid (GlcA) at C5 to iduronic acid (IdoA), O-sulfation at C2 of IdoA and GlcA, C6 of GlcNAc, and C6 of galactose provide a further level of complexity. It is estimated that 3000 GAG determinants are present in the human glycome and an additional 4000 theoretical pentasaccharide sequences are possible. 24 Heparin is the most heterogeneous GAG and contains the highest levels of structural modification, and HS has a related structure but contains areas of high modification interspersed with areas of low modification. HA is the simplest GAG, but unlike all other GAGs, it is not attached to a PG core protein and is nonsulfated. HA is composed of β1–3- and β1–4-linked GlcN and GlcA disaccharides with as many as 10,000 disaccharides assembled to form HA of a molecular size in excess of 5 MDa. HA has a widespread distribution. 25 Despite its relative simplicity of structure, HA is interactive with a diverse range of ECM and cellular proteins, which impact on a number of important physiological processes including wound repair. HA also has high water regain properties and is important in the hydration of tissues providing a highly interactive and hydrated matrix conducive to cell survival and cellular migration. HA is a prominent component of skin and one of its most abundant components with major roles in skin repair (Fig. 4).
Bone repair
After bone fracture, coagulation and inflammatory phases occur in bone similar to those seen in the repair of subcutaneous wounds. A hematoma is initially formed in the fracture site, and fibroblasts and platelets are attracted into the fibrin clot and lay down a transient soft cartilaginous callus with collagen and PGs providing structural support. 26 Biglycan, lumican, aggrecan, perlecan, syndecan-2, and syndecan-4 have been identified in the cartilaginous fracture callus. 27 HA also has roles to play in all stages of fracture repair, particularly in the initial hematoma formation and in the regulation of inflammatory conditions and the influx of cells into the fracture site. Thus, HA, HS, CS, and DS have roles to play in fracture repair, their roles in fracture repair are outlined below. Fracture healing is a complex physiologic process that involves the coordinated participation of several cell types.28–32 Fracture healing can be categorized into direct healing of the fracture site through internal remodeling processes or indirect healing through formation of a transitional callus matrix at the fracture site, which undergoes progressive stages of remodeling to become bone. Like cutaneous wound healing, fracture repair also occurs through a number of identifiable stages. GAGs have roles to play in all stages of the fracture repair process.33,34
Stage 1: hematoma formation.
The first stage is the formation of a hematoma at the fracture site to effect hemostasis, inflammation at the fracture site, followed by the laying down of a soft callus and then a hard callus and remodeling of the fracture repair tissue to produce bone. These stages are represented schematically in Figure 5.
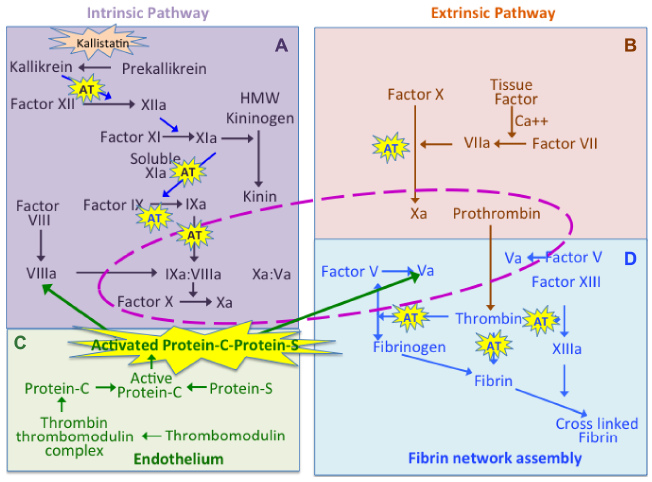
Composite figure depicting the complexity of the coagulation cascade reactions, which regulate blood clot formation in wounds. The interrelationship between the intrinsic pathway (A), extrinsic pathway (B), and events in the endothelium (C) and in fibrin network assembly (D) are shown. Thrombomodulin is a GAG-substituted protein, which has important roles in the regulation of the procoagulant proteins, protein C and protein S. Thrombin and Factor Va have central roles in fibrin network assembly (D). The reactions surrounded by the central dotted pink area in this figure are those which occur on the platelet surface. Key steps in the generation of Xa, Va, and VIIIa in the cascade are modulated by the GAG-regulated serine proteinase inhibitory protein ATIII (AT). Kallistatin is another tissue serine proteinase inhibitory protein regulating the formation of kallikrein, which feeds into the coagulation cascade in early stages of the intrinsic pathway and also regulates the generation of kinins from kininogen, which regulates vasodilation in early stages of wound repair. The APC and protein S are anticoagulant proteins, which regulate Factors Va and VIIIa. Hyaluronan also regulates fibrin assembly in D. ATIII and APC-protein S are highlighted by a yellow flash symbol to highlight their important contributions to the regulation of the coagulation cascade and the key roles they play in wound repair.
Transformation of the hematoma into granulation tissue
After initial hematoma formation over a few hours, inflammation occurs at the fracture site over the ensuing one to seven days. Degranulation of platelets and an influx of inflammatory cells into the fracture repair site occur in a similar manner to that described for cutaneous wound healing. 35 Granulation tissue eventually replaces the hematoma, and osteoclasts remove necrotic bone fragments. Intramembranous ossification then occurs by progenitor cells from the periosteum/endosteum, which differentiates to become osteoblasts forming stabilizing bone collars around the fracture site and filling the medullary canal.
Stage 2: soft callus formation.
On days 7–14 of fracture repair, soft callus eventually replaces the hematoma, ingrowth of capillaries into the soft callus also occurs, and an increased vascular supply to the fracture repair site is also evident. 36 This is an important aspect of the fracture repair process 37 and, as in cutaneous wound repair, is regulated by a number of GAGs. Mesenchymal stem cells migrate into the soft callus and differentiate into fibroblasts and chondrocytes, which produce characteristic ECM components supporting the tissue repair. Thus, the soft callus is converted into a cartilaginous GAG and collagen-rich matrix. A number of PGs support this repair process 38 including syndecan-2, 39 syndecan-4, 40 aggrecan, and biglycan. 41 Syndecan-2 is upregulated in syndecan-4 knockout mice. Wnt-3a upregulates syndecan-2, while TNF-α elevates syndecan-4 levels in vitro. Deficient TNF-α signaling impairs intramembranous bone formation during fracture repair 42 Biglycan and decorin both interact with TNF-α 43 and may have a regulatory role over this inflammatory cytokine in a similar manner to how they regulate TGF-β.44–46 Biglycan also sequesters endostatin promoting the vascularization of the callus repair tissue but also has roles in the formation of the hard callus. 41 Biglycan knock-out mice display an osteo-porotic phenotype, 47 consistent with an increased osteoclast differentiation, a delayed osteogenesis, 48 and an impairment in osteoblast activity 49 demonstrating biglycans regulatory properties in bone formation, remodeling, and repair. 50
Stage 3: hard callus formation.
After the soft callus tissue links the ends of the fractured bone, hard callus formation begins. Intramembranous bone continues to form in the bone collar, the soft callus undergoes endochondral ossification, and the soft callus is gradually converted into rigid calcified tissue. 35 This begins peripherally at the margins of the fracture site where the tissue is under less strain and reduces the strain in a more central location, and this region also undergoes bony callus formation; thus, the fracture repair tissue is replaced by woven bone attached to the original corticol bone. 51
Stage 4: remodeling of the hard callus.
Once the fracture site is replaced with solid woven bone, a slow replacement of this tissue commences with the laying down of lamellar bone, remodeling continues till the bone returns to its original morphology, and the medullary canal is also restored.
Animal models which have been developed to examine the stages of fracture repair and therapeutic interventions to improve repair processes.
The rat52–54 and mouse55–59 are popular models for the examination of a number of aspects of fracture repair. A number of inbred mouse strains 60 have been examined for this purpose as have a number of knockout mice for the examination of a range of biological agents to enhance fracture repair. Rodent models of nonunion have also been developed to examine biological strategies 57 to improve fracture union 61 and the identification of therapeutic targets.
Angiogenesis makes a major contribution to the healing of skin wounds but less so in the healing of relatively avascular tensional and weight-bearing connective tissues such as tendon, intervertebral disk, meniscus, and cartilage. These tissues also have high internal pressure, tough collagenous ECMs, and high PG contents refractory to the penetration of blood vessels, which might otherwise improve healing responses. When these tissues are damaged, the PGs undergo proteolytic processing, and a diminution in their density results in a reduction in the internal hydrostatic pressure and a matrix more conducive to the penetration of blood vessels. The GAG side chains of PGs have antiangiogenic activity, which prevents penetration of blood vessels, and are inhibitory for repair processes in neural tissue.
Cartilage repair
Unlike skin and bone, cartilage does not contain blood vessels. Oxygen and nutrients are delivered by diffusion limiting cartilage's ability to grow and recover from traumatic damage. Small, full-thickness cartilage defects may undergo
Tendon repair
Tendons were once considered to be a comparable tissue to cartilage and inherently incapable of repair due to their avascular nature. 64 However, it is now known that spontaneous healing can occur via proliferation of the epitenon and endotenon or, extrinsically, by invasion of cells from the synovium. Tendons (and bone) show improved healing with a limited level of stress and motion of the defect site during wound repair, emphasizing the stressful environment tenocytes require to maintain optimal function.65–69 Exercise-based rehabilitation programs are designed to accelerate tendon healing and maturational changes to improve the final strength of the repaired tendon. Application of stem cells in tendon repair is also a promising area as is the use of bioscaffolds.66,70,71
Repair of neural tissue
In contrast to the aforementioned tissues where PGs support tissue repair, CS-PGs (lecticans) are refractory to repair in nervous tissues. The CNS and PNS have differing repair potentials. The PNS has an intrinsic capability to regenerate, while the CNS does not. Neurons are the principal functional cell type in the CNS and PNS and represent ~10% of the total cell population, and accessory glial cells (neuroglia,
Neurotransmitter storage and release at the synaptic gap involve GAG interactions
Neurotransmitters are synthesized in the neuronal body rER transported to the Golgi and then to the synapse through a microtubular transport mechanism within the neuron. HS-PGs (agrin and perlecan) and CS-PGs (brevican) act as an ionic intracellular storage media for the neurotransmitters prior to their transfer into synaptic vesicles by synaptic vesicle PG-2, a 12 span transmembrane KS PG with a transporter function, which also immobilizes the neurotransmitters within the vesicles. 100 Small neurotransmitters are stored in small translucent vesicles, while neuropeptides are stored in larger dense vesicles. Neurotransmitters are stored in separate vesicles and not as a mixed population; however, how this selectivity is undertaken awaits clarification but may involve some sorting mechanism by the intracellular CS- and HS-PGs. Neuropeptide transmitters are synthesized as propeptide forms, which are trimmed of a signal peptide prior to transport into the dense synaptic vesicles. The neurotransmitter vesicles are stored in the synaptic terminal until release across the synaptic gap to their respective receptors. This occurs when an action potential depolarizes the presynaptic nerve terminal, voltage-gated Ca2+ (calcium) channels located in the presynaptic terminal membrane open, and Ca2+ ions influx into the synaptic terminal permeability increases. This causes the membrane of the vesicles to fuse with the presynaptic membrane at the active zone and release the neurotransmitter into the synaptic gap to travel to their respective receptors. HS-PGs also have some role to play in the regulation of the generation of this action potential in the neuron and in the coordination of electrical signals in neuronal networks. 101 A deficit of HS leads to an autistic phenotype in mice. 102 Chronic heparinase treatment reduces the mean firing rates of neurons and drastically effects functional maturation of neuronal networks. The action of Sulf1 and Sulf2 in situ may regulate this activity displayed by HS. 100 Reduced levels of agrin and perlecan in the NMJ also detrimentally affect the organization of the NMJ and neuromuscular function.103–105 Brevican also has roles in promoting fast synaptic transmission of signals during neuronal signaling. 106 Members of the syndecan and glypican PG families are also present in the presynaptic gap107–110 where they interact with postsynaptic cell adhesion molecules providing synaptic connectivity and regulate synaptic development, including formation, maturation, and repair, and maintenance of neuronal synapse plasticity.109,111,112
Roles of GAGs in matrix assembly processes relevant to wound repair
GAGs have many and varied roles to play in all stages of wound repair. HA oligosaccharides promote angiogenesis and can synergize with VEGF/PDGF.
113
The HS side chains of perlecan interact with the FGF family, VEGF, PDGF, BMPs, Wnt family, and hedgehog proteins, which promote wound repair.
114
Perlecan sequesters and stabilizes FGF-2 and other members of the FGF family in tissues promoting cellular proliferation, differentiation, ECM synthesis, and tissue development.
115
This is clearly evident in perlecan knockout mice, which have severely impaired large vessel and nerve development,116,117 malformed skull bones and distorted long bones, and severely distorted growth plate cartilages and a short stout stature.
118
Another model has been developed with ablation of a 20 kDa N-terminal portion of the perlecan domain-1 HS attachment sites in
ECM PGs and wound repair
PGs provide architectural support and hydration to tissues conducive to cellular migration into the wound site to promote repair processes (Table 2). Many PGs have a modular design containing functional subdomains important in tissue function. The GAG side chains of PGs have key roles to play in various stages of the wound healing process, and HS interacts and sequesters a number of growth factors at the wound site including FGF family members, as well as BMP, VEGF, and PDGF. These growth factors promote cellular migration, proliferation, differentiation, and ECM synthesis essential for wound healing. The ECM PGs also provide surfaces for cellular attachment and are interactive with a varied number of structural glycoproteins, collagens, and integrins, which stabilize the newly formed wound repair tissue. The lectican PG family forms perineural net structures with HA and tenascin-R in the CNS/PNS. 136 If neural tissues are damaged, repulsive signals from the lectican PGs prevent neural outgrowth and repair processes. Chondroitinase ABC has been used in models, which demonstrate axonal regeneration, identifying the CS side chains as the source of repulsive signals, which inhibit repair.72–74, 78, 137 Table 2 summarizes the major PGs in tensional and weight-bearing connective tissues and their roles in ECM assembly and function. The reader is referred to recent reviews in this subject area for further PG information.11,115,131,138–141
What role do the CS chains on TM play in its anticoagulant activities?
TM acts as a thrombin receptor on the endothelial cell surface. The presence of CS on TM-β decreases the Kd for thrombin binding, significantly accelerating the inhibition of thrombin by AT, and has profound effects on TM's anticoagulant properties.142,143 The C-4-S chains on TM are relatively small (10–12 kDa)144,145 but are essential for its anticoagulant activities. 146 TM-α does not contain a CS chain but contains the HNK-1 linkage saccharide only and is devoid of anticoagulant activity (Fig. 6). In addition to enhancing its cofactor activity for protein C activation, the CS chain on TM-β also affects the inhibition of thrombin-induced fibrinogen clotting, accelerating the inactivation of thrombin by AT.142,147 The CS chain on TM-β binds at least two molecules of thrombin (Fig. 6).
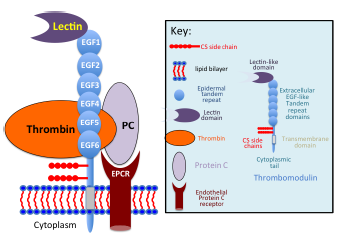
Thrombomodulin (TM), a thrombin receptor, anticoagulant, modulator of inflammation and cell shape. A schematic representation is depicted of the molecular organization and the interactions that occur between thrombin and TM through its EGF domains 5 and 6 and association of protein C (PC) bound to its receptor, EPCR on the endothelial cell surface. This results in the generation of APC, which interacts with protein S also on the cell surface (not shown), and the APC–protein S complex binds to Factors V, VIII, Va, and VIIIa and proteolytically inactivates these. TM-β contains CS chains attached close to its transmembrane domain, which bind thrombin and are essential for its anticoagulant activity. The N-terminal lectin domain of TM interacts with Lewis Y carbohydrate on cells and through these modulates inflammation. Thrombin complexed with TM is rapidly inactivated by antithrombin-III (not shown). TM has six EGF tandem repeats, a transmembrane, and a cytoplasmic domain interactive with the cytoskeletal protein ezrin and associates with actin fibrils, modulating cell shape and focal adhesions affecting cell shape and migration.
GAG interactions with serpins improve their inhibitory properties
GAGs act as cofactors for many of the serpins enhancing their inhibitory properties. The GAG-binding serpins include AT, HCII, and PCI. Heparin and HS bind to AT, HCII, and PCI, and HCII also uses DS as a cofactor. 148 Other serpins such as PAI-1, kallistatin, and α1-antitrypsin also interact with GAGs, accelerating protease inhibition. GAG binding to the serpins, generally occurs to a conserved region on the serpin leading to a conformational change resulting in increased or tighter protease binding accelerating the rates of inhibition up to 10,000-fold compared to the unbound native serpin. 148 Several different AT activities were originally reported in plasma (AT1–IV); however, the major activity was subsequently shown to be due to one molecule, ATIII. The International Society on Thrombosis and Haemo-stasis in 1993 therefore recommended that AT should be used as the standard abbreviation for AT activity. AT is a heparin cofactor and a member of the serine protease inhibitor family (serpin). AT is an important protease inhibitor of thrombin and Factor Xa but can also inhibit IXa, XIa, XIIab, kallikrein, and plasmin and is thus one of the major naturally occurring inhibitors of coagulation. 149
AT is unique among the serpins in that it circulates in plasma in a native inactive conformation. Activation of AT occurs upon binding of domain 1 to a specific pentasaccharide sequence in heparin and HS in the vasculature (Fig. 7).
150
This results in a rearrangement of the reactive center loop of AT in domains 3 and 4 facilitating productive associations with the coagulation proteases. A 3-O sulfate motif in
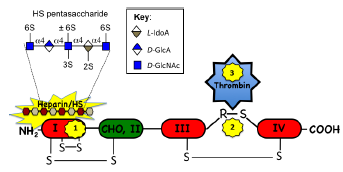
Schematic representation of the structural organization of AT and the molecular rearrangements, which occur to provide protease inhibition. AT is a 52 kDa plasma serpin consisting of four domains linked together by three disulfide bonds, and domain II is glycosylated. A specific pentasaccharide sequence in heparin/HS in the vasculature binds to the N-terminal domain I inducing a conformational change in AT (1) exposing the protease inhibitory site in domains III and IV in the C-terminal half of the molecule (2), which subsequently interacts and inactivates a number of the coagulation proteases (3) (thrombin is shown as a representative example). The specific HS pentasaccharide sequence responsible for interaction with AT is also shown. The 3-O sulfate group on
Heparin also acts as a tight binding competitive inhibitor of human neutrophil elastase (HNE) and also inhibits cathepsin G and mast cell chymase, this is strongly dependent on heparin chain length and charge density. 152 α1-PI, α2-macroglobulin, SLPI, and elafin are also potent inhibitors of HNE, and their inhibitory capacity for the inhibition of HNE is enhanced by HS.153–155 Several serpin members (HCII, PN-1, PAI-1, and PCI) are heparin-binding proteins, and this improves their inhibitory properties. 156 Squamous cell carcinoma antigens (SCCA-1 and SCCA-2) inhibit cathepsin-L through heparin- and HS-mediated interactions, but other GAGs are ineffective. 157 Heparin binding also accelerates the inhibitory properties of tissue inhibitor of metalloproteinase (TIMP)-3, which have a broad inhibitory spectra including interstitial and cell membrane MMPs, and key members of the ADAMs and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) metal-loprotease families. 158 TIMP-3 inhibits MMP-17, an ADAM sheddase. 159 Unlike other members of the TIMP family, TIMP-3 displays limited solubility in free solution and associates with GAG-substituted cell adhesion proteins and PGs localizing TIMP-3 pericellularly in the vicinity of MMPs and related proteinases. TIMP-3 also associates with many proMMP enzyme forms in the PCM, which may improve its inhibitory properties and the protection of the ECM and PCM from excessive proteolysis. 160
Proteinase activation and regulation by GAGs
GAGs have multiple functional properties over proteinases relevant to wound repair (Table 3). Heparin promotes the binding of thrombin to fibrin 161 and also stimulates uPA activity. 162 Heparin also acts as a tight binding competitive inhibitor of HNE and also inhibits cathepsin G and mast cell chymase, and this is strongly dependent on heparin chain length and charge density. 152 Specific cell surface interactions with PGs, integrins, and GAGs have been observed localizing MMPs at the cell surface. 163 These include MMP-2-integrin αvβ3, 164 MMP-9-integrin α4β1, 165 and MMP-1-integrin α2β1. 166 Membrane-type (MT)-MMPs (MMP-14, -15, -16, -17, -24, and -25) are single-pass transmembrane proteins that are active at the cell surface may also act as docking modules for other MMPs and may be actively involved in their activation, e.g, MMP-14 and proMMP-2. 167 TIMP-2 also has a specialized role in the activation of proMMP-2 by MMP-14. The N-terminal domain of TIMP-2 forms an inhibitory complex with the active site of MMP-14, while the C-terminal domain interacts with the hemopexin domain of MMP-2 forming a trimeric activation complex. 168 Cell surface HS-PGs may also act as docking sites for MMPs at the cell surface. 169 Syndecan-2 acts as a docking receptor for proMMP-7 interacting directly with proMMP-7 at the plasma membrane, enhancing its processing into active MMP-7. 170 The major cell surface HA receptor. CD44 may also dock MMP-7 169 and MMP-9 169 to the cell surface. The localization of MMPs at the cell surface confers resistance to TIMP inhibition 171 and promotes their activation in the cell-surface pericellular matrix. ADAMTS and a subset of MMPs, including the MT-MMPs, contain a furin recognition sequence between their propeptide and catalytic domains, and furin convertase enzymes cleave at this site in the Golgi apparatus. 172 Prodo-main removal from MMPs can also be achieved by the action of other MMPs, such as the MMP-14-mediated activation of proMMP-2, or by activation cascades involving coactivator enzymes such as plasmin. 173 Sulfated GAGs also play important roles in controlling MMP activation at the cell surface. MMP-2 and -7 and ADAMTS-5 are shown in Figure 8 as representative examples. HS regulates ADAM 12 through a molecular switch mechanism. The noncovalently associated prodomain in concert with the catalytic domain of ADAM 12 form a novel molecular switch critical for the regulation of the ADAM 12 proteolytic activity by HS-PGs. 174 Via direct interaction with proMMP-7, sulfated GAGs such as chondroitin4,6-sulfate (CS-E) act as allosteric modulators promoting the autolytic activation of the proteinase. Once activated, GAGs may facilitate proteolysis of certain substrates by interacting with the substrate, the enzyme, or both. Activation of proMMP-2 by MMP-16 is also significantly enhanced in the presence of excess chondroitin 4-sulfate (C4S), whereas chondroitin 6-sulfate or low molecular mass HA is ineffective. 175
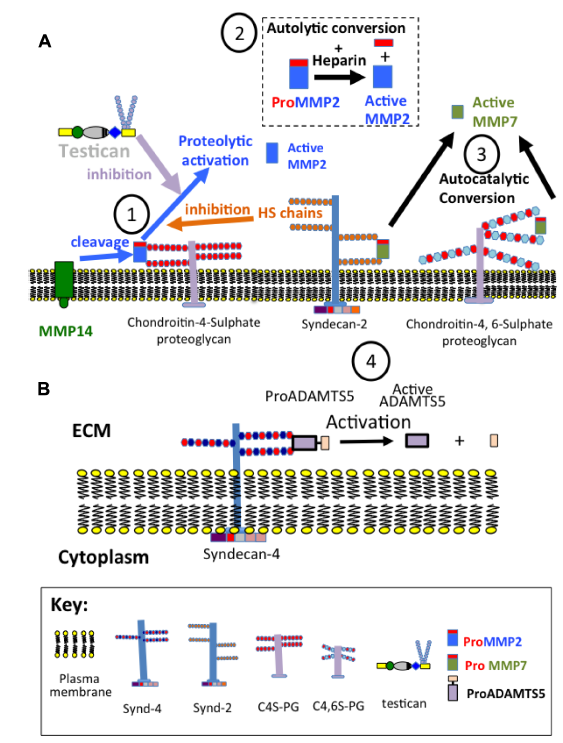
Regulation of MMP activation at the cell surface by GAGs (1–4). MMP-2 and MMP-7 are presented as illustrative examples (A). Heparin promotes autocatalytic conversion of proMMP-2 to active MMP-2. MMP-14 also activates proMMP2 attached to C4S proteoglycan at the cell surface. The HS chains of syndecan-2 and testican inhibit this activation process. The heparan sulphate chains of syndecan-2 and chondroitin-4 and 6-sulphate side chains of cell surface proteoglycans promote autolytic conversion of proMMP7 into active MMP7. The HS chains of syndecan-4 promote the activation of ADAMTS-5.
Subcellular/ECM localization of proteinases and their regulation by GAGs.
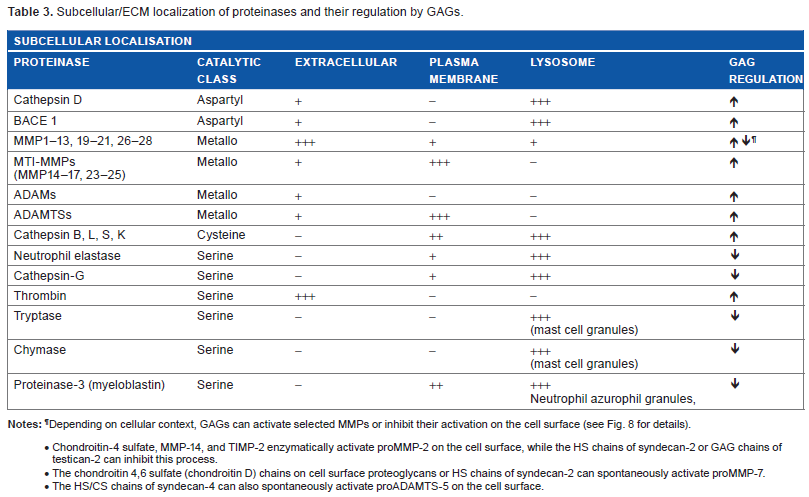
Depending on cellular context, GAGs can activate selected MMPs or inhibit their activation on the cell surface (see Fig. 8 for details).
Chondroitin-4 sulfate, MMP-14, and TIMP-2 enzymatically activate proMMP-2 on the cell surface, while the HS chains of syndecan-2 or GAG chains of testican-2 can inhibit this process. The chondroitin 4,6 sulfate (chondroitin D) chains on cell surface proteoglycans or HS chains of syndecan-2 can spontaneously activate proMMP-7. The HS/CS chains of syndecan-4 can also spontaneously activate proADAMTS-5 on the cell surface.
ADAMTS-5 is the major aggrecanase in mice 176 with an activity at least 1000-fold greater than that of ADAMTS-4 under physiological conditions. 177 ADAMTS-4 activation involves the coordinated activity of both glycosylphosphatidyl inositolanchored MMP-17 and syndecan-1 on the cell surface. 178
Syndecan-4, controls ADAMTS-5 activation through direct interaction with this proteinase. 179 Syndecan-4 is also crucial in the regulation of MMP-3 through the activation of ERK1/2 and by targeting ADAMTS-5 to the cell surface. Thus, loss of syndecan-4 results in a reduced expression of both MMP-3 and aggrecanase activity (Fig. 8). In contrast, syndecan-2 acts as a suppressor for MMP-2 activation on the cell surface. 180 This process involves the HS chains of syndecan-2, and treatment of low metastatic tumor cells with heparitinase-I promotes MMP-2 activation. Another soluble PG, testican, 181 inhibits proMMP-2 processing by MT-MMPs. 182
MMP cascades involving cathepsins (lysosomal cysteine proteinases), which release uPA turn, generate plasmin from plasminogen and the plasmin feeds back into the activation of MMPs illustrating some of the complexities of proteinase activation in situ. Endopeptidases, such as cathepsins B, L, S, and K, can also be activated autocatalytically or by other proteinases such as cathepsin D and pepsin. 183 Lysosomal GAGs interact with and regulate the activities of lysosomal cathepsins. Autocatalytic activation of cysteine cathepsins is substantially accelerated in the presence of GAGs, and these facilitate procathepsin B activation. 184 Furthermore, trafficking of the cathepsins to the cell surface may also involve the action of cell surface HS-PGs. Cathepsin D, an aspartyl lysosomal cathepsin, is also regulated by GAGs. 185 GAGs increase cathepsin D activity in vitro, and heparin increases the activation of cathepsin D from its Pro form. GAGs induce the activation of the β-site amyloid-cleaving enzyme (BACE1), 186 a membrane-anchored enzyme that catalyzes the production of β-amyloid, and its accumulation in the brain of patients with Alzheimer's disease. 187
Heparin and HS stabilize and increase the activity of lysosomal cysteine cathepsins. The mature form of cathepsin B is rapidly inactivated at neutral or alkaline pH and by its endogenous cystatin proteinase inhibitor family, 188 but membrane-bound cathepsin B is very resistant to inactivation at neutral pH possibly through steric effects that prevent the access of TIMPs. 189 Cathepsin K is the principal cysteine proteinase responsible for the degradation of bone. 172 At acidic pH, GAGs expressed in bone and cartilage, such as CS and KS, enhance the collagenolytic and bone resorptive activity of cathepsin K, whereas DS, HS, and HP selectively inhibit its activity; 190 thus, GAGs can participate in the regulation of bone resorption. GAGs act as allosteric modifiers of cathepsin K by influencing its conformational state either enhancing or inhibiting its enzymatic degradative properties. 191
Regulatory molecules in the coagulation cascade and GAGs
GAGs have regulatory roles to play in a number of coagulation proteins, such as TM, protein C, protein S, endothelial protein C receptor (EPCR), and protease-activated receptors 1 and 2 (PAR-1 and 2). Protein S shares homology with other vitamin K-dependent coagulation proteins and acts as a cofactor to protein C in the inactivation of Factors Va and VIIIa. Protein S contains four EGF, one γ-carboxyglutamic acid, and two laminin-G-like domains. Protein C belongs to the peptidase S1 family. It contains two EGF, one glutamic acid, and one peptidase S1 domain. TM (CD141) is a 74-105 kDa cell-surface PG mediator of endothelial anticoagulant activity and activator of protein C. TM contains one N-terminal C-type lectin and six EGF-like domains. It occurs as a part-time CS-PG (β-TM) containing the CS disaccharides –4GlcUAβ1-3GalNAcβ1– sulfated at the C-4 and C-6 positions 192 and a form containing a truncated side chain containing the HNK-1 (HSO(3)–3GlcAβ1–3Galβ1–4GlcNAc–) linkage tetrasaccharide, which is terminated in a 3-O sulfate motif (α-TM).80,192,193 This latter variant form of TM does not display anticoagulant activity. TM is synthesized by endothelial cells, mesothelial cells, monocytes, and a sub-set of dendritic cells. Mutant cells defective in CS elongation do not exhibit anticoagulant activity. The N-terminal lectin-like D1 domain of TM suppresses vascular inflammation by inhibiting leukocyte recruitment to the endothelium by attenuating Lewis Y (LeY)-mediated adhesion. 194 The cytoplasmic domain of TM is bound directly to the N-terminal domain of the cytoskeletal protein ezrin and colocalizes with actin filaments, 195 EGF acting upstream of ezrin activation stimulates the interaction between ezrin and TM maintaining epithelial morphology and cell migration during wound healing.
Multifunctional properties of TM impacting on the coagulation cascade and wound healing
TM is a widely expressed multifunctional cell surface protein, which mediates the activation of protein C and the inhibition of thrombin, generates thrombin-activatable fibrinolysis inhibitor (TAFI), 196 inactivates Va and VIIIa, and has anti-inflammatory activity mediated by its lectin-like D1 domain (Fig. 6). 194 TAFIα is a carboxypeptidase-B enzyme that suppresses fibrinolysis.196,197 Binding of the lectin domain of TM to the cytokine, high mobility group box protein 1, and Lewis Y cell surface tetrasaccharide on endothelial cells provides anti-inflammatory and antiangiogenic activities. 198 Binding of thrombin to TM on the endothelial cell surface prevents the activation of Va by thrombin. Thus, TM acts as an anticoagulant protein through its actions on thrombin and by generation of activated protein C (APC). 143 Once APC is formed, it binds to protein S on the cell surface, and the APC-protein S complex inactivates Va and VIIIa. APC can also act in free solution in the presence of Ca2+, but its action is significantly improved by the presence of phospholipid vesicles.199,200 APC also activates MMP-2 and MMP-9 produced by endothelial cells and stimulates proliferation, migration, and wound closure in cutaneous wounds201–203 and a healing phenotype in cultured tenocytes. 200 APC also activates MMP-2, -9, and -13 produced by chondrocytes in rheumatoid arthritic and osteoarthritic articular cartilages.204,205 Thus, APC modulates ECM remodeling in disease and during repair. 203 TM's multidomain structure and multicomponent interactions with thrombin, protein C, TAFI, complement, Lewis Y antigen, and high mobility group box protein 1, a chromosomal protein regulating transcription, replication, recombination, and DNA repair, facilitates TM's physiologically significant anti-inflammatory, anticoagulant, and antifibrinolytic properties.197,206 APC is inactivated by PCI a widely distrib uted serpin of broad protease specificity or a1-antitrypsin. 207
APC and wound healing
APC has emerging roles as a new biotherapeutic in wound repair applications. 208 APC is the central enzyme in the natural anticoagulant pathway inactivating nonactive and active Factors V and VIII, resulting in an attenuation of thrombin formation and downregulation of coagulation (Fig. 4). The presence of the APC cofactor, protein S, TM, EPCR, and a phospholipid surface contributes to the expression of APC-mediated anticoagulant activity. 209 APC also binds to EPCR in lipid rafts to activate PAR-1 and anti-inflammatory and cytoprotective signaling responses in endothelial cells.210,211 APC also promotes wound healing in chronic ulcers and in bone. 212 Low circulating levels of protein C are associated with lower healing responses in leg ulcers in patients with diabetes. 213 APC increases bone anabolism via a PAR-1/2-dependent mechanism and may synergize with rhBMP-2 for bone repair. 212 APC differentially regulates the viability and differentiation of osteoblasts mediated by bisphosphonates and may be useful in combination therapy for bone regeneration. 214 APC signals through PAR-1 and PAR-2 to activate Akt (RAC-alpha serine/threonine-protein kinase) and increase keratinocyte proliferation and skin wound healing.
Therapeutic application of GAGs and GAG-regulated proteins in wound repair
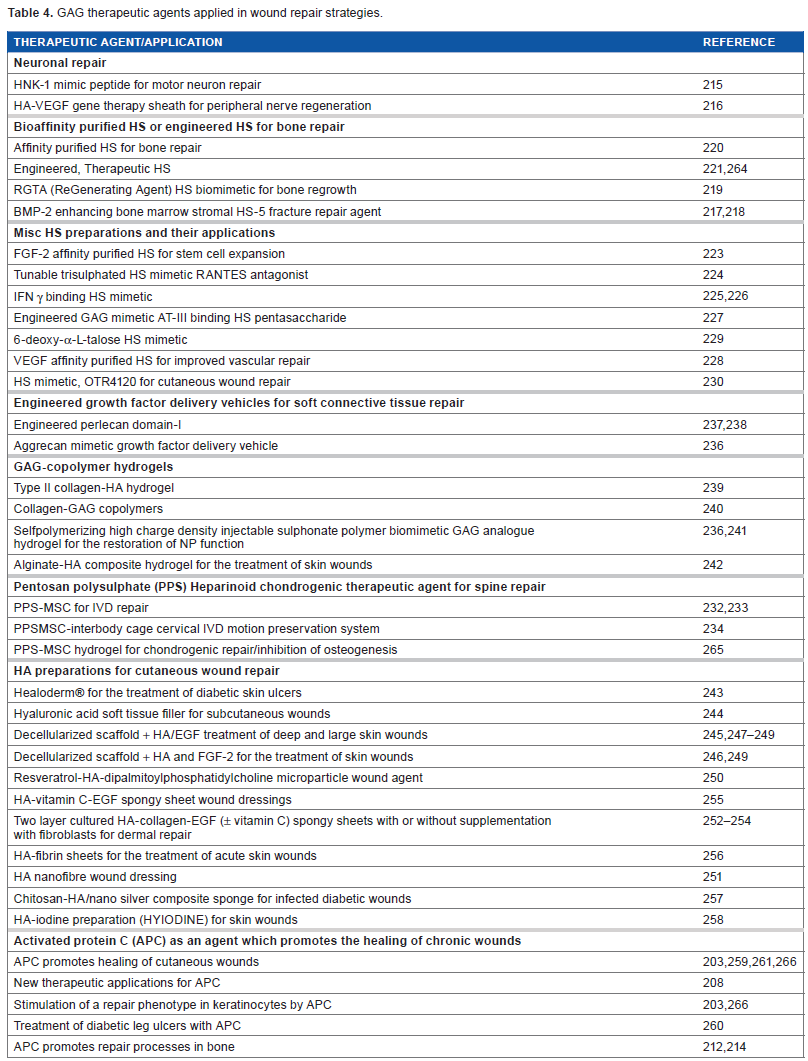
From studies on the pathobiology of the wound repair process and a greater understanding of GAGs in these processes, it has been possible to formulate therapeutic approaches, which improve wound healing. A survey of therapeutic approaches with GAGs, GAG derivatives, or GAG mimetics is presented in Table 4. Peptides have been developed, which mimic HNK-1 to stimulate motor neuron repair. 215 A 93% recovery in quadriceps muscle function was achieved in a three-month recuperative period using this procedure. 215 An HA-VEGF gene sheath has also been developed, which can be placed over damaged axons for peripheral nerve regeneration with minimal scar formation. 216 Heparin and HS are the most extensively modified GAGs and most heterogeneous. Affinity procedures have been developed to isolate HS subfractions for specific therapeutic applications in bone and vascular repairs and in the expansion of progenitor MSCs for regenerative procedures. These include BMP-2 interactive HS,217,218 RGTA biomimetic HS, 219 affinity-purified HS, 220 and engineered HS samples 221 for enhanced bone repair. HA-GDF-5 hydrogels have also been developed for bone repair. 222 FGF-2 affinity-purified HS for enhanced expansion of stem cells, 223 a trisulfated HS RANTES antagonist, 224 IFN-γ-binding HS,225,226 AT-binding HS pentasaccharides, 227 and VEGF affinity-purified HS 228 for an improved vascular repair. HS biomimetics based on 6-deoxy-α-talose, 229 OTR4120 mimetic HS 230 for an improved cutaneous wound repair, and HS biomimetic clusters 231 have been developed. The heparinoid pentosan polysulfate has found application in IVD232,233 and cartilage repair and in combination with MSCs and spinal interbody cages 234 for spinal fusions. Engineered aggrecan235,236 and perlecan domain-1 growth factor delivery vehicles237,238 have been developed, and GAG-copolymer hydrogels for wound repair include HA-type II collagen, 239 collagen–GAG, 240 high-charge density sulfonate analog GAG biomimetic for IVD restoration, 241 and alginate–HA composite hydrogels. 242 A diverse range of HA formulations and products have been developed to treat cutaneous wounds including Healoderm® for the treatment of diabetic ulcers, 243 HA soft tissue fillers, 244 decellularized scaffolds derivatized with HA/EGF,245–249 and HA/FGF-2246,249 for the treatment of deep large skin wounds. Resveratrol–HA microparticles, 250 HA nanofiber dressings, 251 HA ± vitamin C-EGF spongy dressings supplemented with dermal fibroblasts,252–255 and fibrin–HA sheet dressings 256 have been applied for the treatment of acute and chronic skin wounds. Chitosan–HA/slow release nanosilver composite sponges 257 and HA–iodine preparations (Hyiodine™) 258 have been used for the treatment of infected diabetic wounds. Application of APC in the treatment of diabetic wounds offers a particularly novel treatment in a clinically demanding area of chronic skin wound repair.203,208,259–261 APC may also prove useful in combination therapy with bisphosphonates or BMP-2 in the treatment of bone lesions. 214
GAG therapeutic agents applied in wound repair strategies.
Conclusion
A greater understanding of the roles that GAGs play in tissue homeostasis and wound repair has fueled significant advances in treatments for the repair of clinically demanding wounds in neural tissues and diabetic ulcers. The application of MSCs for tissue repair in tensional and weight-bearing tissues has also yielded promising results, and GAGs have essential regulatory roles in the proliferation and differentiation of progenitor cell populations, which undertake wound repair. The sophistication of GAGs as an information delivery system to cells is significant and capable of directing many aspects of cellular behavior, which control tissue homeostasis and wound repair. A greater understanding of the glycocode provided by GAGs will further advance the treatment of chronic and acute wounds.
Abbreviations
TGF-β, transforming growth factor beta; IGF-I, insulin-like growth factor-I; IL-1, interleukin-1; TNF-α, tumor necrosis factor alpha; PDGF, platelet derived growth factor; FGF-2, fibroblast growth factor-2; IFN-γ, interferon gamma; MMPs, matrix metalloproteinases; NCAM, neural cell adhesion molecule; NgCAM, neuroglia cell adhesion molecule; NMJ, neuromuscular junction; MSCs, mesenchymal stem cells; ADAMS, A disintegrin and metalloproteinase; ADAMTSs, A disintegrin and metalloproteinase with thrombospondin motifs.
Author Contributions
JM Conceived the study, analyzed the data, and wrote the manuscript.