
Abstract
Cisplatin is a known antitumor drug, but its mechanisms of action are not fully elucidated. In this research, we studied the anticancer potential of cisplatin at doses of 1, 2, or 3 μM using HL-60 cells as a test model. We investigated cisplatin effects at the molecular level using RNA sequencing, cell cycle analysis, and apoptotic assay after 24, 48, 72, and 96 hours of treatment. The results show that many genes responsible for molecular and cellular functions were significantly altered. Cisplatin treatment also caused the cells to be arrested at the DNA synthesis phase, and as the time increases, the cells gradually accumulated at the sub-G1 phase. Also, as the dose increases, a significant number of cells entered into the apoptotic and necrotic stages. Altogether, the data show that low doses of cisplatin significantly impact the viability of HL-60 cells, through modulation of gene expression, cell cycle, and apoptosis.
Introduction
Cisplatin, cis-diamminedichloroplatinum (II), is an organometallic compound that has antiproliferative properties and induces apoptosis in many cell types. It was approved as the first platinum-based anticancer drug in 1978, and this led to the exploration of other platinum (II)- and metal-containing anticancer drugs.1,2 It has been clinically proven that cisplatin acts against various human cancers, and it has been used for the treatment of lung cancer,3,4 ovarian cancer,5,6 carcinoma,7,8 breast cancer,9,10 brain cancer, 11 bladder, esophageal, and head and neck cancers. 12 Another novel therapeutic strategy that has been applied for cancer treatment is to treat the cancer cells with cisplatin along with other cancer or noncancer drugs. Paclitaxel and cisplatin combination has been used to treat breast cancer, ovarian cancer, and melanoma.13–15 Other drugs including doxorubicin, 16 gemcitabine, 17 vitamin D, 18 and other natural compounds like osthole, honey bee venom, and anvirzel have been used with positive outcomes in various cancer treatments.5,19,20
Unfortunately, in addition to benign anticancer properties, cisplatin has various side effects on normal renal, sensory, hair, and neuron cells. Additionally, most of the cisplatintreated cancer cells exhibit relapse in response to the drug during the later stages due to drug resistance because of probable changes in drug intake, biotransformation, or DNA repair mechanisms. 21 Since its clinical importance has been discovered, from the past four decades, research has been conducted to unravel the molecular mechanism of action. Broadly, cisplatin is known to bind DNA and form DNA adducts, which is aimed to subsequently inhibit cell cycle progression and induce apoptosis.22,23
In the present study, we investigated molecular mechanisms of cisplatin pharmacology at lower doses of exposure. We used promyelocytic HL-60 human leukemic cell line as test model since they have been reported as a good model of drug response studies at molecular level. 24 Our goal is to evaluate the low-dose cisplatin effect at the molecular level. With this goal, previously we reported the oxidative stress, lipid peroxidation, and DNA damage in HL-60 cells in response to 1, 2, or 3 μM of cisplatin. In the present study, we studied gene expression profiles, cell cycle analysis, and apoptosis status of the cells at similar conditions and their relevance to the previous findings from our laboratory. 25
Methods
Cell culture
The HL-60 cells were obtained from the American Type Culture Collection and incubated in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) from Thermo Fisher Scientific and penicillin/streptomycin. The cultures were incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
RNA sequencing
The treated (1, 2, or 3 μM of cisplatin) or untreated HL-60 cells (n = 3) were incubated for 96 hours. After incubation, total RNA was isolated from the cells using RNAqueous Total RNA Isolation Kit from Life Technologies. The isolated total RNA samples were analyzed at the Molecular and Genomics Core Facility of University of Mississippi Medical Center. To monitor the samples' processing progress, we have entered the sample information into laboratory information management system. The isolated RNA samples were column purified using PureLink™ RNA Mini Kit (Invitrogen), and the quality of the RNA was evaluated (Bio-Rad Experion System). Quality-tested samples with minimum concentration and size range were used to develop RNA libraries (n = 12) with TruSeq Stranded Total RNA with Ribo-Zero Kit Set A (Illumina). Each sample was processed with a total of 1 μg RNA and the resulted cDNA was quantified by Qubit system (Invitrogen). The quality and size of the cDNA libraries were assessed with Experion DNA 1K chip (Bio-Rad) according to the manufacturer's instructions. The cDNA libraries were generated with fragment sizes that ranged from 220 to 500 bps with a peak at 260 bps. The generated libraries with 10 nM concentration were stored at −20 °C, and the rest of the libraries were diluted to 2 nM. A total of 10 μL of 2 nM libraries were denatured and sequenced using NextSeq 500 High Output Kit (300 cycles – PE100) on Illumina NextSeq 500 platform. To evaluate the sequencing reads, they were automatically uploaded and evaluated using Illumina BaseSpace Onsite Computing platform. FASTQ sequence files were generated and preliminary analysis was carried out with the applications available on BaseSpace Onsite Computing platform, including TopHat Alignment (read mapping to reference genome-UCSC-hg19) and Cufflinks Assembly & DE (assembly of novel transcripts and differential expression). GeneSifter™ software platform (http://www.genesifter.net) was used for the additional analysis. Ingenuity Pathways Analysis (Ingenuity® Systems, www.ingenuity.com) software was used to evaluate gene networks and functional analysis. 26
Cell cycle assay
Treated (1, 2, or 3 μM of cisplatin) or untreated HL-60 cells were incubated for 24, 48, 72, or 96 hours in 5% CO2 at 37 °C. After the incubation, cells were collected by centrifugation and washed twice with Phosphate-Buffered Saline (PBS). The positive control HL-60 cells were treated with 20 μM arsenic trioxide. Both treated and untreated cells were fixed with 70% ice cold ethanol on ice for 30 minutes, washed twice with PBS, and incubated with PI/RNAse staining solution from Nexcelom Bioscience according to the manufacturer's instructions. The stained cells were analyzed using a Cellometer, and Cellometer vision CBA software from Nexcelom Bioscience.
Apoptosis
Apoptotic properties of the treated or untreated cells were analyzed using Cellometer from Nexcelom Bioscience according to the manufacturer's instructions. HL-60 cells were treated with cisplatin (1, 2, or 3 μM), Arsenic Trioxide (ATO) (20 μM), or left untreated. After 24, 48, 72, or 96 hour incubation, the cells were washed with PBS and the pellets were dissolved in Annexin V binding buffer followed by incubation with Annexin V-FITC and propidium iodide (PI; Nexcelom Bioscience) according to the manufacturer's protocol. The stained cells were analyzed using Cellometer and Vision CBA software from Nexcelom Bioscience.
Statistical analysis
Statistical analysis was performed using Student's t-test and a P-value of <0.05 was considered as significant. Differentially expressed genes were evaluated using t-test [P < 0.05 and fold change ± 1.5 or greater] by two methods: (1) FWER and (2) Benjamini and Hochberg false discovery rate, which corrects for multiple comparisons.
Results
Effects of cisplatin on gene expression in HL-60 cells
To study early genes response to low doses of cisplatin drug in HL-60 cells, the cells were untreated or treated with 1, 2, or 3 μM of cisplatin for 96 hours and subjected to RNA sequencing. The analysis yields a list of genes for each treatment compared to the corresponding controls at P-value <0.05, and the list of genes for each test concentration is presented in Supplementary Tables 1–3 for the concentrations 1, 2, and 3 μM, respectively. The expression of almost 37 genes was altered significantly (P < 0.05) in response to 1 μM of cisplatin compared to the control (Supplementary Table 1), whereas in 2 and 3 μM treated cells, more than 1000 gene expressions were altered significantly (P < 0.05; Supplementary Tables 2 and 3). A total number of 28 genes were consistently altered in 1, 2, and 3 μM of cisplatintreated HL-60 cells (Table 1). Since the 28 genes consistently altered at all the tested cisplatin concentrations, we focused more on the analysis of those 28 genes that are upregulated or downregulated significantly compared to the controls (Table 1). Among the 28 genes, 22 genes were upregulated and the rest of the six genes were downregulated. The upregulated genes induced E2F transcription factor 1 (E2F1), E2F transcription factor 2 (E2F2), E2F transcription factor 8 (E2F8), IQ motif containing GTPase activating protein 3 (IQGAP3), vimentin (VIM), DNA damage-inducible transcript 4 (DDIT4), family with sequence similarity 111, member A (FAM111A), growth arrest-specific 7 (GAS7), thymidine kinase 1, soluble (TK1), anti-silencing function 1B histone chaperone (ASF1B), ribonucleotide reductase M2 (RRM2), V-myb avian myeloblastosis viral oncogene homolog-like 2 (MYBL2), interleukin 8 (IL8), transcription factor 19 (TCF19), tubulin, alpha 4a (TUBA4A), RecQ protein-like 4 (RECQL4), Mov10 RISC complex RNA helicase (MOV10), Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 24 (SLC25A24), Fanconi anemia, complementation group G (FANCG), interleukin 27 receptor, alpha (IL27RA), cyclin E1 (CCNE1), and maternal embryonic leucine zipper kinase (MELK), whereas the downregulated genes are cell division cycle 20 (CDC20), disks, large (Drosophila) homolog-associated protein 5 (DLGAP5), PIF1 5′-to-3′ DNA helicase (PIF1), karyopherin alpha 2 (RAG cohort 1, importin alpha 1) (KPNA2), breast carcinoma amplified sequence 3 (BCAS3), and histone cluster 1, H2bl (HIST1H2BL).
List of cisplatin-altered genes in HL-60 cells.

All the listed genes were either directly or indirectly associated with cell cycle, cellular assembly and organization, DNA replication, recombination, repair, gene expression, or cancer network functions. Since most of the genes were associated with cell cycle, apoptosis, and its related functions, cell cycle analysis and apoptosis assays were performed.
Dose- and time-dependent effects of cisplatin on cell cycle distribution in HL-60 cells
Cisplatin-treated HL-60 cells were arrested in cell cycle progression (Fig. 1). At lower dose (1 μM) and 24 hours of cisplatin treatment, the cells started to accumulate in sub-G1, S, and G2 phases. As the treatment time increases from 48, 72, and 96 hours, the cell distribution was significantly altered in all the cell cycle phases compared to the control cells. Almost 40% of the cell density was reduced at G0/G1 phase, reduced in 1 μM treated cells, and accumulated in S and sub-G1 phases. Interestingly, 2 and 3 μM of cisplatin-treated cells significantly accumulated at sub-G1 phase of the cell cycle with an increase of 30% and 51% cells, respectively. The cell cycle analysis indicated that, at lower doses of cisplatin treatment, the cells were arrested at sub-G1, S, or G2 checkpoints, and as cisplatin dose and treatment time increases, the cells started accumulating more in sub-G1 phase. Accumulation of cells in sub-G1 phase of cell cycle in PI treatment indicated that the cells may be undergoing apoptosis/necrosis.
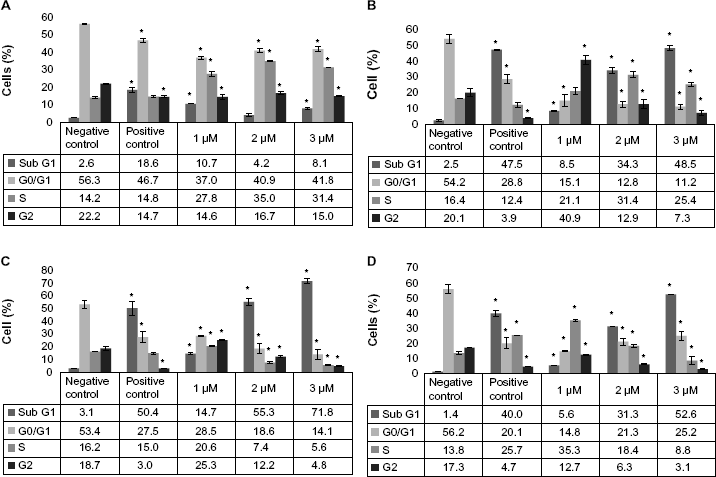
Cisplatin modulation of cell cycle progression in HL-60 cells. HL-60 cells were kept untreated (control) or treated with 1, 2, or 3 μM of cisplatin for 24, 48, 72, or 96 hours. Post treatment, the cells were fixed and incubated with PI and analyzed using Cellometer Vision as described in the “Methods” section. Each graph represents the cell cycle data for 24-hour (
Cisplatin induces apoptosis/necrosis in HL-60 cells
To further analyze the cells for apoptotic/necrosis properties, the cisplatin-treated cells were stained with PI and Annexin V. Annexin V specifically binds phosphatidylserine in cell membrane in which the cells entered into apoptotic phase, whereas PI stains DNA in necrotic cells where the cell membrane disintegrates. The results show that cisplatin induced apoptosis/necrosis in HL-60 cells in a time- and dose-dependent manner. In 24-hour treatment, at doses of 1, 2, or 3 μM, apoptosis was induced by approximately 7%, 10%, or 12% compared to the control cells, respectively (Fig. 2). As the incubation period increases (48, 72, or 96 hours), apoptotic/necrotic cells increased proportionally with the doses. At the end of 96-hour treatment, apoptotic/necrotic cells were significantly increased in all the tested doses compared to the controls (Fig. 3). These results support the evidence that the accumulation of cisplatin-treated cells in sub-G1 phase of cell cycle is due to the cells entering into apoptotic or necrotic phases.

Cisplatin induced apoptosis/necrosis in HL-60 cells. HL-60 cells were untreated or treated with 1, 2, or 3 μM of cisplatin for 24, 48, 72, or 96 hours. The treated and untreated cells were incubated with Annexin V and PI in annexin buffer, and the cells were analyzed using a Cellometer Vision as described in the “Methods” section. The four quadrants of each unit represent the status of the cell population. The top left quadrant represents the damaged cell population, top right quadrant represents the necrotic cell population, bottom left quadrant represents the live and healthy cell population, and bottom right quadrant represents the apoptotic cell population.

Percentages of apoptosis and necrosis in HL-60 cells exposed to cisplatin. HL-60 cells were kept untreated (control) or treated with 1, 2, or 3 μM of cisplatin for 24, 48, 72, or 96 hours, incubated with Annexin V and PI, and analyzed using Cellometer Vision as described in the “Methods” section. The results are presented in bar graphs. Each graph represents the data for apoptosis/necrosis for 24-hour (
Discussion
Various reports on the anticancer properties of cisplatin have been published, but the actual mechanisms of action are largely unknown. Specifically, data for cisplatin effects at lower doses are scarce. In this study, we investigated the effects of low doses (1, 2, and 3 μM) of cisplatin on gene expression in HL-60 cells following 96 hours exposure and determined their relevance to the major cellular and molecular events such as cell cycle modulation and apoptosis. The RNA sequence analysis allowed us to find the list of genes that are altered for each dose (1, 2, or 3 μM; Supplementary Tables 1, 2, or 3) and the list of genes that are consistently altered in all the tested concentrations (1, 2, and 3 μM; Table 1). We narrowed down our analysis and focused more on the 28 genes that were consistently upregulated or downregulated in response to 1, 2, and 3 μM cisplatin (Table 1).
All of the transcription levels of the 28 genes were altered in a dose-dependent manner with higher expression levels being found at the higher doses (2 or 3 μM) compared to those at the lower dose (1 μM). Interestingly, compared to 2 or 3 μM doses, only 3.8% genes' expressions were altered significantly in 1 μM cisplatin-treated cells. Among the 28 genes that were altered consistently in all the tested doses, E2F2 expression levels were strongly upregulated with almost 1-fold change in lower dose and 1.74-fold change in higher dose. It is interesting that E2F2 expression level was previously reported to play a pivotal role in ovarian cancer and suggested to be a potential therapeutic target.27,28 The most downregulated genes were PIF1 and BCAS3. Both genes were repressed by almost 1-fold at lower dose and 1.75-fold at higher doses (Table 1). BCAS3 expression levels have been reported to be higher in cancer cells,29,30 but the reduced transcription levels in response to cisplatin in a dose-dependent manner indicate that the drug has potential even at lower doses of treatment. Similarly, PIF1 targeted siRNA, reduced transcription levels, inhibited cell cycle progression, and elevated apoptosis in colon cancer.31–33 The dose-dependent expression shows an alteration of more genes at higher doses, which directly or indirectly controls the cell proliferation. However, the functional end point of genes regulated at different levels including transcriptional, translational, and posttranslational levels is different. In this report, the dose–response was studied at the transcriptional level. However, further research is needed to evaluate the potential effects of cisplatin at the translational and posttranslational levels.
The RNA sequencing analysis of cisplatin-treated HL-60 cells shows that the CDC20, E2F1, E2F2, E2F8, GAS7, TK1, MYBL2, TCF19, CCNE1, and MELK genes play a major role in cell cycle progression. CDC20 is a known coactivator of anaphase-promoting complex (APC) that interacts with various other proteins in cell cycle regulation.27,28,34–36 APC is responsible for mitotic exit by cyclin destruction. However, the APC-CDC20 complex activity is dependent on the activity of cyclin–CDK activity.37,38 Cyclin is a partner of CDK, as the cyclin activity is repressed, CDK activity decreases and facilitates APC-CDC20 to exit the cell from the mitotic phase. However, the cisplatin-treated cells show decreased levels of CDC20 and increased levels of one of the cyclin (CCNE1) mRNA levels (Table 1). For both genes, either increased cyclin activity with CDK complex or decreased CDC20 levels with APC complex might favor the cells not to exit the mitotic phase. Accordingly, our results show that the cell cycle progression is blocked (Fig. 1) due to either decreased levels of CDC20 or increased levels of CCNE1. It has been reported that the expression levels of cyclins influence cisplatin effect on ovarian cancer cells, 39 endometrial carcinoma cells, 40 and mitotic phase of the cell cycle. E2F family transcription factors are important in cell cycle regulation and control of antiproliferating proteins. Our results show that the E2F1, E2F2, and E2F8 mRNA levels were upregulated in response to cisplatin treatment (Table 1). In consistence with the current results, it was recently reported that E2F contributes significantly in cisplatin-induced cell death in ovarian cancer.41,42 TK1 mRNA levels were significantly induced in a dose-dependent fashion. TK1 is a known cell cycle regulator enzyme, more significantly in S phase.43,44 In fibrosarcoma tumor model, TK1 protein levels were reported to be modulated in response to cisplatin treatment. 45 The current results show that TCF19 and MYBL2 genes expression levels were induced in response to all the tested cisplatin doses. However, the product of TCF19 gene is known to function during G1/S transition, whereas MYBL2, also known as b-Myb, is a known player in cell cycle regulation. These studies underscore the role of these proteins in the cell cycle regulation. 46 In addition, MYBL2 is a known biomarker for cervical cancer, and altered levels of this gene in response to cisplatin have significant relevance to the cancer disease. 47
The cell cycle analysis results are in consistence with the mRNA expression profiles in response to the cisplatin (Table 1). Cisplatin-treated cells were arrested in G0/G1 phase in a dose-dependent fashion. Also, we reported previously that the same doses of cisplatin induced DNA damage in HL-60 cells in a time- and dose-dependent manner. 25 These results indicated that cisplatin induces DNA damage and influences the cell cycle checkpoints to arrest the cells at a particular cell cycle phase. Growth arrest and DNA damage-inducible protein 45 (GADD45a) plays a significant role in DNA repair and suppression of cell growth.48,49 In consistence with the previous reports, the RNA sequencing data of the present study indicate that GADD45a mRNA was induced in 2 and 3 μM cisplatin-treated cells by 0.45 and 0.64 folds, respectively. The arrest of the cell cycle progression could be a result of induced expression of GADD45a and other key cell cycle regulatory proteins in response to cisplatin treatment. Similar results were reported previously indicating that cisplatin induces GADD45a through extracellular signal-regulatory kinase pathway and regulates cell cycle checkpoints.50,51 PIF1 transcription levels were decreased consistently in a dose-dependent manner (Table 1), PIF1 is reported to play a major role in unwinding of replication fork during replication, repair, and transcription. Depletion of PIF1 has been reported to impair the genome-wide replication fork, 52 indicating that cisplatin successfully inhibits the cell cycle progression by decreasing the transcription levels of PIF1. Another protein KPNA2, upregulated in a dose-dependent manner, is known to carry nuclear localization signal for checkpoint protein Chk2, which is responsible for multiple checkpoint arrest in cell cycle phases. 53 Moderately upregulated gene expressions of FAM111A, FANCG, and RECQL4 are reported to play a role in DNA replication, 54 homologous recombination, 55 and mitochondrial genome stability, 56 respectively. The results are consistent with the previous reports indicating that gene expression plays a role in cell cycle checkpoints and DNA repair mechanisms.
Further, we studied the cisplatin potential for apoptosis in HL-60 cells. Cisplatin significantly induced apoptosis in a time-dependent fashion (Figs. 2 and 3). The apoptotic potential of cisplatin is supported by the cisplatin-induced mRNA profiles presented in Table 1. GAS7 gene products are known to control growth arrest and apoptotic property of cells in response to stimuli. As reported earlier, GAS7 gene knockdown reduced apoptosis induced by cisplatin, whereas overexpression of GAS7 gene activated apoptosis. 57 In consistence with the previous reports, the GAS7 gene mRNA levels were upregulated in cisplatin-treated cells in a dose-dependent fashion. Similarly, MELK mRNA levels were upregulated in response to cisplatin in a dose-dependent manner. MELK gene product is also known to play a role in apoptotic induction. It has been reported that overexpressed levels of MELK reduced the survival of breast cancer patients. 58 The other mRNA profiles, including BCAS3 that is overexpressed in cancer cells, were downregulated in all the cisplatin-treated cells in a dose-response fashion, whereas DDIT4 mRNA levels were upregulated. DDIT4 is known to be induced in cisplatin-treated cells in response to reactive oxygen species. 59
Cisplatin has been used for the treatment of a variety of human cancers including testicular, ovarian, bladder, breast, cervical, stomach, head and neck, prostate, esophageal, sarcoma, and neuroblastoma.60–62 However, the current research findings including genes' transcriptional alterations (Table 1), inhibition of cell cycle progression (Fig. 1), and induction of apoptosis (Figs. 2 and 3) in HL-60 cells give new insights into further therapeutic development of cisplatin as a potential drug for leukemia. These results are in consistence with our previous reports on cisplatin-induced oxidative stress and DNA damage. 25 Studies on the cytotoxic mode of action have also revealed that cisplatin induces cell proliferation inhibition, DNA adducts formation and oxidative stress, cell cycle arrest, and apoptosis in HL-60 cells. 63 Additional reports by Previati et al and Floros et al pointed out a similar potential of cisplatin as a possible chemotherapeutic drug in HL-60 cells, by inducing apoptosis, inhibiting cell cycle progression, and modulating oxidative stress.64–66 However, cisplatin doses in these studies are several orders of magnitude higher than the ones we tested in the present investigation. Hence, testing the chemotherapeutic property at low doses offers the advantage of reducing the chemical toxicity, reducing cost, and improving the benefits of treatment. Interestingly, most of the 28 genes that responded to cisplatin treatment in the current study are relevant biomarkers that have been reported in cisplatin-based treatment of other cancer diseases.
Conclusion
In conclusion, we have reported the transcriptional modulation of gene expression in response to cisplatin treatment of HL-60 cells by RNA profiling. Also, we have found that cisplatin induces cell cycle arrest and apoptosis in a dose- and time-dependent manner. Cisplatin significantly altered the expression of many important genes in HL-60 cells, and 28 genes were consistently altered in all tested doses (1, 2, and 3 μM). Interestingly, the genes that were modulated at the transcriptional level were directly or indirectly related to functional pathways and/or end points such as cell cycle arrest, apoptosis, DNA damage, and oxidative stress responses. However, further research is needed to study the specific mechanisms of these biologic responses at the transcriptional level as well as translational level of upregulated or downregulated genes. It is likely that this process may lead to novel and improved therapeutic approaches.
Author Contributions
Conceived and designed the experiments: VV, SRD, PBT. Analyzed the data: VV, SRD, PBT. Wrote the first draft of the manuscript: VV. Contributed to the writing of the manuscript: SRD PBT. Agreed with manuscript results and conclusions: VV, SRD, PBT. Jointly developed the structure and arguments for the paper: VV, PBT. Made critical revisions and approved the final version: VV, PBT. All the authors reviewed and approved the final manuscript.