
Abstract
Recognizing an increasing need for biomarkers that predict clinical outcomes in type 1 diabetes (T1D), JDRF, a major funding organization for T1D research, recently instituted the Core for Assay Validation (CAV) to accelerate the translation of promising assays from discovery to clinical implementation via a process of coordinated evaluation of biomarkers. In this model, the CAV facilitates the validation of candidate assay methods as well as qualification of proposed biomarkers for a specific clinical use in well-characterized patients. We describe here a CAV-driven pilot project aimed at identifying biomarkers that predict the rate of decline in beta cell function after diagnosis. In a formalized pipeline, candidate assays are first assessed for general rationale, technical precision, and biological associations in a cross-sectional cohort. Those with the most favorable characteristics are then applied to placebo arm subjects of T1D intervention trials to assess their predictive correlation with beta cell function. We outline a go/no-go process for advancing candidate assays in a defined qualification pipeline that also allows for the discovery of novel predictive biomarker combinations. This strategy could be a model for other collaborative biomarker development efforts in and beyond T1D.
Introduction
Type 1 diabetes (T1D) occurs when an autoimmune response against the beta cells of the pancreatic islets leads, over a period of anywhere from months to decades, to reduced insulin production capacity and inability to properly utilize dietary sugars. In common with other autoimmune diseases, T1D has a clear hereditary component. Disease risk is linked to multiple genes, the strongest of which is the human leukocyte antigen (HLA) locus, underscoring the central role of the T cell response in the disease.1,2 The earliest signs of islet autoimmunity are the appearance of beta cell antigen-specific autoantibodies, 3 frequently occurring in the first year of life. The number of autoantibody specificities present is highly predictive of onset.4,5 Thus, autoantibody status can identify subjects with a defined risk for developing the disease within a certain period of time and has been used successfully as an eligibility criteria in secondary prevention trials, which aim to delay the progression from islet autoimmunity to diabetes. 6 It is also possible to conduct primary prevention studies, where interventions are given prior to autoantibody development, by identifying at-risk subjects based on genetics. This approach has been made feasible through broad population-based HLA typing at birth, as was used in large studies in Finland, Germany, and the United States.7–9 Thus, both genetic and antibody-based biomarkers, sometimes in combination, have been very successful in the prevention space. However, these biomarkers have not shown the same utility in the postdiagnosis period. Specifically, there is a critical need to understand correlates of the rate of beta cell loss after diagnosis and to predict which subjects are likely to respond to immune therapies.
There has been a growing appreciation that coordinated validation efforts could accelerate the development of biomarkers, helping to move them from the discovery stage to correlation with key clinical outcomes, and then through assay validation for use in clinical trials. In response to these challenges, the JDRF Biomarker Working Group (BWG) was formed in late 2012 to accelerate the development of clinically applicable biomarkers in T1D. The idea to establish a collaborative research group was formalized at a JDRF-organized workshop and previously reported. 10 One feature of the BWG is the Core for Assay Validation (CAV), which has been formed to aid in this process. The CAV aims to coordinate specific biomarker discovery and validation projects by serving two primary functions: (1) ensuring the technical quality of assays used in defining biomarker associations, thus maximizing their statistical power and (2) defining the clinical correlations of multiple biomarker assays analyzed in composite, thus taking into account the likely multifactorial nature of heterogeneity within T1D patients.
Of course, T1D is not the only field that has used coordinated validation and qualification efforts to identify biomarkers associated with important clinical outcomes. For example, samples from the relatively limited number of infected subjects in the human immunodeficiency virus RV144 vaccine trial were distributed based on a preplanned pilot analysis performed for a large number of assays.11,12 Assays were included in or excluded from the project based on technical and biological variability criteria. Immune biomarkers significantly associated with infection risk were identified at the end of the study. The assay development pipeline outlined by the CAV for its first biomarker identification project follows many of the same logical lines.
C-Peptide Prediction Project
The operational functions of the CAV are represented in the C-peptide Prediction Project, the first CAV-facilitated biomarker effort. The goal of this project is to identify biomarkers that, singly or in combination, can be used to predict the rate of disease progression at the time of T1D diagnosis. This type of biomarker could be used as entry criteria for new-onset trials designed to achieve efficacy endpoints with fewer subjects.
C-peptide induced by a mixed meal tolerance test (MMTT) is currently the standard primary endpoint measure in recent-onset T1D clinical trials. 13 C-peptide is a cleavage product of proinsulin during its normal physiological processing and is secreted alongside insulin. C-peptide levels, therefore, correlate with endogenous insulin production but are not affected by administration of exogenous pharmaceutical insulin and, unlike insulin levels, are less affected by first-pass metabolism in the liver. At the time of T1D diagnosis, stimulated C-peptide levels are greatly reduced relative to normal, corresponding to reduced pancreatic beta cell mass, and there is substantial variation between individuals in the rate of C-peptide decline in the initial years after diagnosis. Some subjects will maintain residual insulin secretion for decades, while others will stop producing measurable levels of C-pep-tide within a few years. 14 Those who maintain insulin secretion capacity longer have fewer T1D-associated complications. 15 Adults diagnosed with T1D are more likely to have residual C-peptide production than people diagnosed as children with similar disease durations. 14
The heterogeneity in rates of loss of C-peptide not only impacts healthy outcomes for patients but also impacts clinical trial design. In randomized placebo-controlled new-onset trials, inclusion of slow progressors, or subjects whose C-peptide does not significantly decline over the course the trial, adversely affects the likelihood of detecting an intervention effect, thereby increasing the required trial size. The potential reasons for this are both biological and statistical. Especially in the case of immunomodulatory therapy, slow progressors may not have a significant ongoing autoimmune response to modify, and so are not likely to benefit. Second, regardless of whether a therapy has the intended biological effect on these subjects, there is not enough change in their clinical outcome – change in C-peptide production before and after therapy – to be detectable via the primary endpoint. For both these reasons, the ability to use biomarkers to identify subjects with a higher likelihood of significant C-peptide decline would allow for smaller, faster clinical trials in patients who are more likely to benefit from therapy.
In order to identify predictive biomarkers of C-peptide decline, the CAV has assembled multiple candidate assays for inclusion in the C-peptide Prediction Project. These assays, based on either preliminary data or other a priori information, are hypothesized to predict C-peptide fall in the first two years after diagnosis. The current assays are spread across multiple categories, including assessment of islet-specific T cell frequencies, beta cell death and dysfunction, serum microRNAs, and circulating cytokine profiles. Additional assays are being explored to fill other biological “spaces” such as Treg signatures and B cell phenotypes. The specific assay designs, analytes, and performance characteristics will be reported in the future by the contributing investigators as the data are generated. The CAV is also performing support assays on the sample sets in order to give insight into the relationship of each biomarker outcome with underlying patient biology. These support assays, including ribonucleic acid sequencing (RNAseq) of both whole blood and fractionated peripheral blood mononuclear cell (PBMC) subsets, multiparameter fluorescence-activated cell sorting immunophenotyping, and serum cytokine measurements, will provide a comprehensive baseline profile of donor immune status against which to interpret other biomarker measurements.
Biomarker Qualification Process
The C-peptide Prediction Project represents the first iteration of a biomarker qualification pipeline that could be applied to multiple clinical questions. The pipeline is outlined in Figure 1 and contains three initial steps: (1) a priori knowledge of the assay, (2) results from blinded replicate testing, and (3) biological variability data from a cross-sectional T1D cohort. These three criteria will be combined into a single scorecard for each assay, which will be used to select assays ready to receive well-characterized samples from clinical trials. The specific criteria for go/no-go decisions at each step in the process are described below. The ultimate goal of this work is to qualify biomarkers for correlation with future C-peptide decline, ideally culminating in Food and Drug Administration (FDA) recognition of their utility as a Drug Development Tool suitable for use in both industrial and academic clinical settings. The C-peptide Prediction Project represents the first major step in this pipeline. In general, the process focuses on biomarker qualification, that is, identification of the biological meaning and clinical utility of the characteristic(s) being measured. In the long term, we expect that assays with solid clinical correlations will undergo full assay validation process that is appropriate for commercialization and use as a Drug Development Tool.
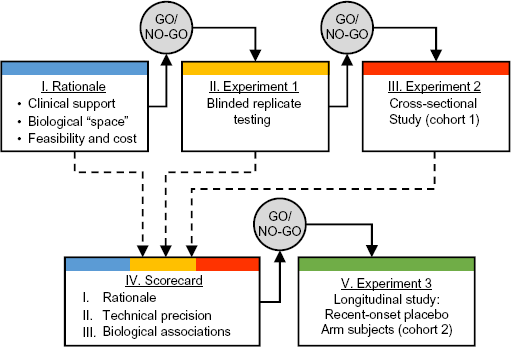
Validation pipeline for C-peptide prediction project. A priori knowledge of the assay, results from blinded replicate testing, and biological variability data from a cross-sectional T1D cohort will be assessed for each assay. These three criteria will be combined into a single scorecard for each assay as indicated by the dashed lines. Highest ranking assays will receive samples from recent-onset T1D clinical trials.
We have made several strategic decisions in establishing this pipeline. For example, a preliminary assessment of assay precision, via blinded testing of replicate samples, is included to ensure that assays meet a minimum bar for performance to merit inclusion in downstream analyses. Rather than focusing on in-depth assay optimization early, we recommend that more extensive characterizations of assay performance and standard operating procedure development efforts can be reserved for biomarker assays initially demonstrating clinical utility. The system is designed to identify biomarkers that are detectable in specific sample types, collected by specific methods used in T1D clinical trials. Assays that do not pass all pipeline steps may be useful in other contexts (eg, a different clinical question) or using samples of different quantity or quality (eg, fresh instead of frozen). Others have successfully used workshop approaches to advance assays toward standardized use in T1D; certainly, this is not the only possible method for assay evaluation. This pipeline can serve as a starting point, and can be adjusted for use in other settings.
Here, we outline our proposed process for C-peptide predictive biomarker qualification.
Rationale for inclusion
Each assay, whether derived from the research community or contributed by the CAV as a core measure, is evaluated and advanced via the process described in Figure 1, beginning with an assessment of the rationale and feasibility characteristics of the assay. All assays included in the C-peptide Prediction Project were screened to determine whether they represented a unique “biological space” or whether they were duplicative of biomarkers already included in the project. We evaluated the clinical support for each assay, such as previous associations with clinical parameters like disease duration, or whether T1D and healthy subjects were known to have different quantities of the analyte. Assays were also assessed for feasibility criteria such as sample volume (as defined by sample sizes available from the final C-peptide decline cohort) and cost. Assays requiring sample sizes that were too large, that were duplicative of other efforts, or that had no previous findings in T1D were not advanced to further steps in the pipeline.
Blinded replicate testing
The first laboratory-based step is an evaluation of the technical precision of the assay to ensure a minimum level of reliability of the outcome, using defined samples provided by the CAV. Precision is assessed by having the investigator measure, in a blinded fashion, three or more biological replicate aliquots from the same blood draw from five or more subjects (Fig. 2). For each assay, appropriate statistical measures of variability are calculated, most commonly the coefficient of variation (CV). The CV standard selected for this pilot project was 30%. Assays with CVs >30% can repeat replicate testing or modify their assays to improve precision; the CAV can help with either of these possibilities, but assays will not advance to Cohort 1 without acceptable CV characteristics.

Blinded replication as initial validation step. Each assay is issued triplicate aliquots from a single blood draw from five individual subjects. These 15 samples are assayed in a blinded fashion. CAV staff unblind the data and calculate CV for each subject. Some assays will have multiple analytes assessed in this same manner.
For assay platforms that measure multiple parameters at once, both the assay as a whole and individual analytes within the assay are evaluated in this fashion. For example, RNAseq was performed on triplicate aliquots of frozen PBMC sorted to obtain monocytes, among other cell types. Sample thawing and cell sorting occurred on different days for each aliquot, and there are multiple steps during the sample preparation procedure, which are potential sources of technical variability. Despite this, we identified over 5000 transcripts that are detected with high consistency (CV <10%) across the three monocyte samples, as well as a smaller subset (~1000) that are detectable in monocytes but highly impacted by technical factors (CV >30%). These latter imprecisely quantified transcripts will be excluded from further analysis, but the assay as a whole will continue through the pipeline using only the robustly detected transcript data in downstream statistical models.
Cross-sectional study (Cohort 1)
Assays with a sufficient number of individual analytes demonstrating acceptable precision (CV ≤30%) will advance to the next step: analysis of a cross-sectional cohort of T1D subjects. These subjects have a wide range of disease characteristics, including age at diagnosis, duration of disease, and C-peptide level (Fig. 3). All the subjects have a broad array of matched samples available so that multiple biomarker assays can be run in combination. This pilot cohort will serve as an initial dataset from which to draw associations with clinical parameters as well as between biomarker assay types (eg, beta cell death and T cell frequencies). Assays that correlate strongly with disease duration or C-peptide levels at draw will be prioritized, but these correlations are not required for inclusion in further pipeline steps. These data will also be used as an initial measurement of biological variability, which should be relatively high when looking at a broad set of subjects. Markers that do not vary significantly between individuals are less likely to be useful and will not move forward through the pipeline. Finally, from a practical standpoint, we will look at assay success/failure rates, or how often data are returned for each sample distributed, to ensure basic assay reliability prior to distributing clinical trial samples. Assays must be sufficiently reliable in order to pass this go/no-go step.

Cross-sectional cohort of T1D subjects is clinically varied. Subjects in the cross-sectional cohort have a range of disease durations (x-axis), and ages at diagnoses (y-axis). Some subjects in this cohort continue to produce C-peptide after diagnosis (X-marks).
Data synthesis – scorecard
A combined assessment of characteristics from each of the first three evaluation steps (rationale, technical precision, biological associations) will be made via an aggregated scorecard. Assays which rank most highly in the scorecard system will advance to Cohort 2 and will receive samples to address the primary experimental objective of the C-peptide Prediction Project.
Longitudinal study of recent-onset subjects (Cohort 2)
Cohort 2 is composed of samples from subjects randomized to the control or placebo arms of recent-onset T1D clinical trials. Samples currently in use for the project come from three trials sponsored by the Immune Tolerance Network, a National Institute of Allergy and Infectious Diseases-sponsored clinical research consortium. 16 The AbATE, 17 START, 18 and T1DAL 19 trials all had similar designs, enrolling individuals within three months of diagnosis of T1D. Longitudinal mechanistic samples (PBMC, serum, whole blood RNA) were banked at the 0, 6, and 24-month visits, as well as other timepoints along the course of the trials. In addition to providing samples for cryopreservation, trial subjects complete extensive clinical characterization, including baseline and follow-up MMTT-stimulated C-peptide [two-hour area under the curve (AUC)] data, which in all cases was the primary trial endpoint. This C-peptide outcome will serve as the dependent variable to which we will draw biomarker correlations. This will allow us to determine which assay or combination of assays best predicts the change in C-peptide AUC over two years (Fig. 4).

Decline in C-peptide is variable in clinical trial subjects. C-peptide decline in subjects from placebo arms of ITN's published AbATE 17 and START 18 trials are depicted here. Data are colored by tertile% reduction in C-peptide production. Blue: subjects with slowest decline in C-peptide (“slow”). Red: subjects with most rapid decline in C-peptide (“rapid”). Green: intermediate decline in C-peptide.
All assays will be performed in a blinded fashion by participating laboratories. After data capture, the sample identities are unblinded and data are incorporated into a “training” dataset that will be used to define baseline classifiers of rapid versus slow progression. Data will be analyzed by statisticians at the CAV and will also be made available to the contributing laboratory. In order to minimize penalties due to multiple testing, CAV analysis will be conducted on analytes meeting strict criteria. The primary analysis will involve modeling a limited number of highly precise analytes as selected via the scorecard system. The maximum number of analytes included in the primary analysis will be calculated based on the size of Cohort 2, the technical variability of the assay as determined by blinded replicate testing, and the estimated minimum effect size for association with C-peptide outcome. This strategy will give us the highest likelihood of identifying significant correlations. Secondary exploratory analyses will be possible involving analytes meeting less stringent requirements.
Future Steps
Recent unsuccessful efforts to replicate published cancer biomarker studies highlight the need for better prequalification and replication studies. 20 In our training dataset, we expect at least a small subset of biomarkers to correlate with C-peptide loss, either alone or in a combinatorial fashion. Either way, they will need to be repeated in an independent cohort to confirm their clinical association. There is an urgent need to identify additional sources of recent-onset T1D subject samples with associated longitudinal clinical outcome information. We are currently exploring other existing clinical trial sample banks and, in parallel, planning additional prospective recent-onset cohort studies for this purpose.
Biomarker assays that are proposed for multicenter trial use should also show repeatability in other laboratories and conditions (also known as robustness), although in our view this type of assessment is more appropriate later in the development pipeline. For some assays, transfer to another laboratory could be accomplished with relative ease. For example, qPCR-based assays directed at one or a few messenger RNAs or miRNAs should be testable at many institutions, requiring limited specialized equipment and commonly held laboratory skills. These and similar assays could be tested at the CAV to ensure transferability of protocols and reagents prior to seeking good laboratory practice (GLP) validation. For assays that require specialized equipment or skills, it would be advisable to determine if they can be miniaturized or simplified prior to GLP development efforts. Either way, considerations about the current state of the detection assay should not be a barrier to investigate the correlations of the biomarker itself, with the assumption that the contributing investigator, the CAV, and future commercial partners could all potentially contribute to any assay optimization needed for continued development.
Summary
The CAV is promoting one avenue for collaborative biomarker evaluation by providing a framework for investigators to work toward assay validation and qualification for a defined clinical purpose. We have developed a pipeline for multiassay interrogation of common clinical sample sets in a systematic manner and are developing robust analytical methods to handle multidimensional biomarker datasets. The C-peptide Prediction Project represents the first CAV-organized effort employing these formalized methods to identify biomarkers that could have a major impact on the conduct of T1D trials. Our hope is that this project will serve as a template for collaborative biomarker discovery and qualification both within and beyond the field of T1D clinical research.
Footnotes
Acknowledgments
Research involving human subjects discussed in this manuscript was approved by the Benaroya Research Institute Institutional Review Board. We wish to thank Dr. Simi Ahmed from JDRF for programmatic guidance and as well as members of the JDRF Biomarker Working Group for being essential contributors to the C-peptide Prediction Project and many other BWG efforts not discussed here. Samples used for these studies come from participants in the BRIDGE study, conducted at the Benaroya Research Institute by Dr. Carla Greenbaum and colleagues, and from the Immune Tolerance Network, to whom we are grateful. Connor McCoy and Kelly Geubtner assisted with the figures for this manuscript. We also thank Dr. Johnna Wesley for inviting us to submit this article.
Author Contributions
Wrote and revised the manuscript: CS, JO. Reviewed and approved the final manuscript: CS, JO.