
Abstract
Infectious diseases affect human health despite advances in biomedical research and drug discovery. Among these, viruses are especially difficult to tackle due to the sudden transfer from animals to humans, high mutational rates, resistance to current treatments, and the intricacies of their molecular interactions with the host. As an example of these interactions, we describe a cell-based approach to monitor specific proteolytic events executed by either the viral-encoded protease or by host proteins on the virus. We then emphasize the significance of examining proteolysis within the subcellular compartment where cleavage occurs naturally. We show the power of stable expression, highlighting the usefulness of the cell-based multiplexed approach, which we have adapted to two independent assays previously developed to monitor (a) the activity of the HIV-1-encoded protease or (b) the cleavage of the HIV-1-encoded envelope protein by the host. Multiplexing was achieved by mixing cells each carrying a different assay or, alternatively, by engineering cells expressing two assays. Multiplexing relies on the robustness of the individual assays and their clear discrimination, further enhancing screening capabilities in an attempt to block proteolytic events required for viral infectivity and spread.
Keywords
Introduction
Viral–Host Cross Talk and Proteolysis
Viral infection has affected human health throughout history. The difficult, long, and expensive race to bring new drugs to the market is further convoluted by the appearance of new viral diseases rapidly spreading around the world. Among the most recent emerging viral pathogens, chikungunya virus,
1
Middle East respiratory syndrome coronavirus,
2
and Zika virus,3–5 to mention a few, are of great concern to the World Health Organization. For example, the recent spike in Zika virus infection in several states of Brazil has health officials scrambling to understand the specific aspects of this virus's pathogenicity.6–8 While vaccine development is under way and Zika–dengue virus cross-reacting antibodies have been described,9–12 antibody-dependent enhancement will need to be addressed.
13
Thus, as part of the efforts of the scientific community to alleviate the
Understanding the viral life cycle and its discrete steps is a requirement for targeted discovery. Furthermore, taking into consideration the specific location in the cell where each step occurs can enhance the power of targeted drug discovery that will ultimately lead to potent and efficient antivirals. This is particularly so with proteolysis, as proteolytic events occur in various subcellular compartments.26,27 Monitoring cleavage in these compartments should elucidate the cellular requirements for cleavage in a natural cellular
HIV-1 PR Assay
HIV-1 PR has been one of the best-characterized and studied viral-encoded proteins, driven mostly by the HIV-1/AIDS pandemic and the need for antivirals. It also exemplifies a viral protein active within the cytosolic environment of the viral capsid at the late stages of the viral life cycle prior, during, and post budding.31,32 HIV, the causative agent of AIDS, is a lentivirus within the
The viral aspartyl PR is responsible for the posttranslational cleavage of the viral proteome, cleaving all but one site.20,38,39 It is also known to possess autocatalytic properties as it cleaves itself from the precursor Gag-Pol polyprotein when two molecules dimerize before processing the rest of the viral proteome. 40 The host enzyme furin processes the boundary between gp120 and gp41 of Env. 20
The assay we have previously described to monitor PR activity relies on the well-characterized Gal4 yeast transcription factor broadly utilized in the classical two-hybrid system and for regulated gene expression. The assay, similar to that in the two-hybrid system, exploits the N-terminal DNA-binding domain and the C-terminal transactivation domain of Gal4. However, in the assay, the HIV-1 PR is fused between the two domains of Gal4 and (when PR is mutated or inhibited) activates transcription of the enhanced green fluorescent protein (eGFP) gene downstream the Gal4 upstream activation sequence (UAS). 28 In such a way, the assay can easily discriminate between active and nonactive PR based on fluorescence and was engineered in an inducible manner to circumvent possible cytotoxic effects of PR when expressed in mammalian cells. 41
HIV-1 Env Assay
As mentioned, the only HIV-1 protein not cleaved by the viral PR is Env, which is cleaved by furin in the vesicles of the classical secretory pathway, primarily in the Golgi apparatus. 42 The classical secretory pathway represents an essential system for the posttranslational modification and transport of proteins to the cell surface. Constituted of the ER, Golgi, and trans-Golgi network, the classical secretory pathway houses a multitude of enzymes responsible for posttranslational modifications including signal peptidases and signal peptide peptidases that are responsible for cleaving off signal sequences for ER targeting,43,44 the clearing of misfolded proteins from the ER, 45 and the maturation of certain viral proteins during infection, most notably HCV core protein.46,47 Additionally, a family of enzymes known as proprotein con-vertases are responsible for the removal of inhibitory domains, leading to the activation and maturation of certain proteins. 48 HIV-1, for example, utilizes this pathway for the cleavage/ maturation of its viral-encoded Env protein en route to the cell surface. Once translated, the Env glycoprotein gp160 is translocated to the ER where it is posttranslationally modified to its mature and active form by the prototypic proprotein con-vertase furin, a process absolutely required for the production of infectious virions.49–51 The Env subunits gp120 and gp41 are then transported to the cell surface where they remain noncovalently associated and are incorporated into budding viral particles. 42
Previously, we described an assay that was designed to specifically monitor the proteolytic processing of HIV-1 Env in the classical secretory pathway. The assay relies on a double-tagged scaffold fusion protein engineered to localize to the ER and traverse the classical secretory pathway en route to the cell surface. At the cell surface, the scaffold is embedded within the cell membrane. Cleavage of the gp120/gp41 boundary, or lack thereof, is recognized by antibody staining and detected by classical methods such as flow cytometry. This allows us to discriminate in an accurate and robust manner between cleaved and uncleaved events based on the presence of one and two tags, respectively. 29
Multiplexing and Genetic Barcoding
Through the years, multiplexing has become an attractive tool for biomedical applications to enhance the power of analysis. Combining high experimental efficiency with decreased cost, multiplexing has proven a practical method for both in vitro studies and cell-based approaches.52,53 Defined as the simultaneous evaluation of several experimental elements, multiplexing can be applied to a wide variety of systems and formats in flow cytometry54–59 or microscopy. 60 Specifically, our laboratory has successfully combined the use of fluorescent proteins and genetic engineering through retroviral technology to obtain cell lines in order to generate a variety of multiplexed formats. Furthermore, this allows us to ensure stable, nondeteriorating expression of different fluorescent proteins at different intensity levels.61,62 The utility of such formats was demonstrated by Stolp et al who used different cell-based platforms genetically barcoded for high-throughput screening purposes.29,30
Here, we show additional multiplexing capabilities by combining two biological systems, each one interrogating an independent process: one that monitors cleavage by the viral-encoded protease and the other that accurately detects cleavage by the host enzyme furin. Additionally, we demonstrate the usefulness of genetic barcoding for high-throughput screening (HTS) applications and the robustness and reliability associated with such techniques. We highlight the importance of selecting clonal populations stably expressing the assays. We also compare a mixture of cell populations each expressing an assay, with a population of engineered cells carrying the two assays. Our results further illustrate the power of multiplexing and genetic engineering through retroviral technology. Focusing here on proteolytic cleavage (by the virus and/or the host), this strategy enhances our ability to monitor the viral-host interactions of a broader spectrum of viruses and/or proteins. Proteolytic processing carried out in different manners by various proteins can be evaluated in an elegant and rapid manner. This provides a valuable tool for researchers to learn new viral behaviors not well understood as well as the potential to discover novel protein–protein interactions that have yet to be characterized.
Results
Combining Systems: Testing Two Biological Applications by Mixing Them
Previously, we have shown our ability to robustly and accurately monitor proteolytic cleavage by both the HIV-1 PR and HIV-1 Env boundary platforms.28,29 Although the two assays are based on fluorescence, they are very different in nature. The first relies on eGFP fluorescence (when PR is inhibited), and the second on classical fluorescently labeled antibodies (presence of the second tag when cleavage is inhibited; Fig. 1). Antibody staining allows for flexibility as one can use an array of fluorescently labeled antibodies (primary or secondary). We thus investigated the possibility of exploiting the distinct fluorescent features of the two assays to analyze them in combination. For that purpose, we utilized allophycocyanin (APC)-labeled secondary antibodies against the HA tag for the second assay, as APC's emission spectrum does not overlap with eGFP.
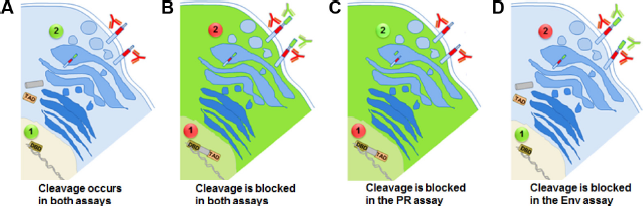
Depiction of the
The Power of Genetic Engineering: Transient Expression versus Stably Expressing Cell Lines
Although transfection experiments are useful for high levels of protein expression and detection, their transient nature leads to the eventual loss of expression of the gene of interest. The rate of this decline is dependent on several key factors including the protein half-life, the stability of the protein, and the rate of cell division of the transfected cells. 63 For many years, retroviral technology has provided a solution for this loss of gene expression.64–68 Through retroviral technology and fluorescence-activated cell sorting (FACS), a distinct cell population can be easily amplified based on the appropriate fluorescent phenotype.69,70 Our goal is to ultimately obtain cell lines expressing two independent assays to provide a robust tool for the screening of antivirals. It is thus critical that we first prove these assays can be expressed independently and then tested in conjunction, whether by mixing cells or in the same cell background. In order to do so, we first performed transfection experiments including all necessary elements of the assay and then proceeded to amplify selected sorted populations with the goal of obtaining stable cell lines expressing each engineered assay. We proceeded with the first assay for monitoring HIV-1 PR, which is much more complex than the second that monitors cleavage of HIV-1 Env as it relies on three plasmids rather than one (refer to “Materials and Methods” section for details). Cells were induced with doxycycline (Dox) and analyzed for inhibition in the presence of the PI darunavir (DAR) as observed by the presence of eGFP-positive cells. As expected and shown in Figure 2, there is a drastic increase in eGFP-positive cells after several rounds of flow cytometry analysis, as compared to the original co-transfection experiments. This illustrates the usefulness and importance associated with engineering stably expressing cell lines, particularly when they are intended for further use in screening and/or multiplexed applications.
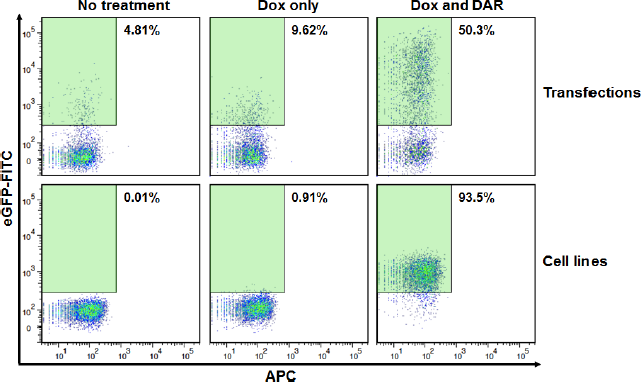
Comparison of transient protein expression in mammalian cells versus stable expression in cell lines. Top panels: HEK 293T cells were transfected with all necessary assay components and then treated with 1 µg Dox and/or 1 µM DAR and analyzed for eGFP expression. Bottom panels: As above but with stably expressing clonal cells. The percentage of eGFP expressing cells within the green gates is shown in each case.
Figure 3 shows that each of the individual assays depends on different fluorescence-based detection methods and can be clearly distinguished from each other. The individual PR assay shows a robust shift to the fluorescein isothiocyanate (FITC) channel (eGFP positive following DAR treatment; top panels). The individual Env assay shows a robust shift to the APC channel (HA positive; bottom panels). Importantly, when combined by mixing them at equal ratios, the mixed population depicts the expected phenotype at the anticipated rate of approximately 50%. When the mixed population is treated with the PI DAR, a shift of approximately half of the population to FITC positive is observed (Fig. 4A, green window). Similarly, when stained with anti-HA antibody, a shift of approximately half of the population to APC positive is observed (red window). These results clearly illustrate that each assay can be distinguished within the mixed population based on the specific treatment or staining conditions required for each system. The gated FITC positive (green population) and APC positive (red population) can be combined and yet be clearly distinguished (Fig. 4B).
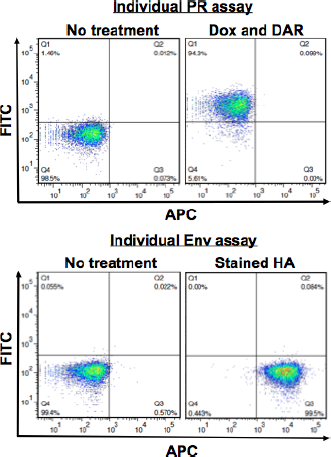
Illustrating the difference in fluorescence-based detection between assays. Human SupT1 cells clonal for both previously described assays (HIV-1 PR and HIV-1 Env) were analyzed for their respective activity. Top panels show the individual PR assay untreated cells (left panel) or treated with 1 µg Dox and 1 µM DAR (right panel). The bottom panels depict cells harboring the individual Env assay either unstained (left panel) or stained with anti-HA antibody (right panel).
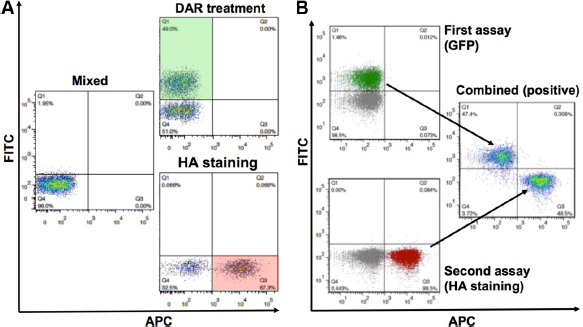
Multiplexing two cell-based platforms for enhanced biological applications. Human SupT1 cells clonal for each developed assay were mixed and analyzed by appropriate fluorescence-detection methods to demonstrate multiplexing capabilities. (
Combining Systems: Testing Two Biological Applications by Combining Them in the Same Cell
Combining samples by mixing them and differentiating between multiple assay readouts is useful but not necessarily special or novel.71,72 It remains to be seen that a cell line harboring two independent assays can be used as a platform where each one of the assays can be de-convoluted or distinguished from each other. Therefore, we proceeded to establish within the same cell the two biological systems we have previously developed in individual cells. For that purpose, a cell line expressing the assay for the HIV-1 PR, and thus all the elements necessary (the rtTA, the UAS-eGFP reporter, and the Gal4/PR fusion), was transduced with retroviral particles carrying the HIV-Env assay construct. Figure 5 depicts the flow cytometry analysis of cells that were genetically engineered to express both assays. The first panel in Figure 5A shows the negative controls (untreated and unstained, without or with Dox). Following induction with Dox and treatment with the PI DAR, a large percentage of cells turn positive for eGFP (green window). In order to prove that these cells carry the Env assay as well, the cells were stained with HA antibody and APC-coupled secondary antibody to corroborate that the scaffold protein traveled to the cell surface. When stained for HA-APC, a large percentage of cells turns positive for HA (red window). Importantly, a large percentage of the population turns positive for both eGFP and HA (double-positive population in the purple window) when cells are both stained for HA-APC and induced and treated with DAR. In order to further demonstrate that the Env assay monitors Env clevage in these cells, the cells were treated with the furin inhibitor decanoyl-RVKR-chloromethylketone (DCK) and stained with FLAG and APC-coupled secondary antibody (Fig. 5B). A similar single-positive eGFP population (green window) appears in FLAG-APC-stained cells following Dox and DAR treatment. More importantly, a similar single-positive (red window) or double-positive population (purple window) appears following DCK treatment. This clearly demonstrates that these cells robustly express both assays simultaneously and can monitor PR activity, Env cleavage, or both. These results prove that each system can clearly and distinctly be monitored based on the treatment required for the detection of each.
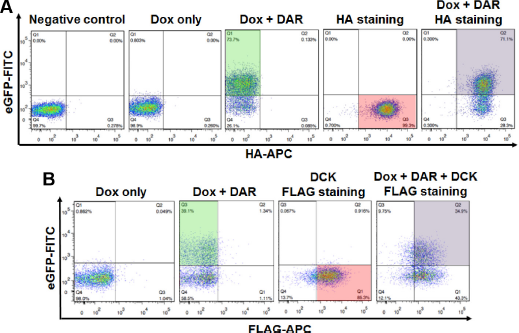
Combining two biological systems within the same cell. Utilizing retroviral technology and FACS, SupT1 cells were engineered to express both PR and Env assays. Cells were analyzed by flow cytometry following each treatment and their combination. (
Discussion
Cell-based approaches are increasingly seen as a valuable tool in molecular biology and drug development. It is becoming increasingly obvious that better and more robust cell-based assays are required for phenotypic drug screening and particularly for targeted screening. The painful path to drug discovery emphasizes the need for pinpointed cell-based assays that interrogate specific processes and/or targets at the very early screening stages.
Cell-based platforms, as the ones we have developed, are a critical and reliable tool for enhancing early-stage drug discovery. Due to their cellular nature, they provide a straightforward approach that circumvents the need for independent or additional cytotoxicity testing, or drug intake through cell membranes, thus reducing the cost and time at these critical initial stages of drug discovery. The cellular nature also provides a better milieu for investigating biological processes such as protein-protein interactions or discerning the factors required for the full activity of the enzymatic function under investigation.
The platform we have established focuses on proteolytic cleavage, a critical set of processes in the intricate relationship between pathogen and host required for the establishment of viral infection. As part of this platform, we have previously developed a cell-based assay that monitors proteolytic cleavage by the viral-encoded protease, which was successfully implemented for HIV-1 PR. We have also developed an assay that monitors cleavage within the classical vesicles of the classical secretory pathway. The assay was engineered to specifically monitor the cleavage of the HIV-1 Env gp120/gp41 boundary by the host enzymes residing in the Golgi/trans-Golgi network, most notably furin.
Here, we have exploited the power of cell line engineering to further enhance our abilities to study viral-host interactions through a targeted approach that focuses on proteolytic cleavage. We also intended to strengthen the capabilities of antiviral screening by creating cell lines, demonstrating that such technologies can be exploited for distinct biological applications as long as they rely on different readout parameters. Utilizing retroviral technology for genetic engineering, we showed a dramatic increase in assay robustness in stable cell lines when compared to transient expression experiments. Stable cell lines should drastically increase assay sensitivity, thus allowing for improved investigative efforts and more reliable high-throughout drug screening.
Additionally, we have shown the power of combining biological applications that monitor/test the proteolytic activity or cleavage of different viral proteins. This was possible due to the different fluorescent nature of each individual assay. The assay that monitors the HIV-1 PR activity relies on eGFP. The assay that monitors cleavage of HIV-1 Env relies on fluorescently labeled antibody staining and thus can use any fluorescent channel distinguishable from the fluorescence channel of eGFP (FITC). Here, we have thus tested whether mixed cell populations harboring each respective assay could still distinctively display its specific phenotype, giving us the ability to monitor activities of multiple targets simultaneously. Analysis by flow cytometry clearly showed that the mixed populations of cells, tested following their individual treatment (induction with Dox and treatment with PI for the PR assay, and HA staining for the Env assay), did indeed display their appropriate phenotype (eGFP expression and HA surface expression, respectively). Our results clearly demonstrate that different biological systems can be combined, provided each system is robust enough and distinguishable from its counterpart and accurate readouts can be obtained from each respective assay.
We then proceeded to investigate whether the two assays can still be distinguishable in the same cell background rather than in mixed cell backgrounds, each expressing one assay. In order to combine the two assays, additional genetic engineering efforts were required. Exploiting retroviral technology to express the Env assay within a cell line originally expressing only the elements of the PR assay, we successfully generated a stable cell line harboring both established assays. The engineered cell line was analyzed for each respective system without the need of accurate mixing of two distinct populations. Our results showed robust readouts for each individual assay. This was the case whether cells were treated for the PR assay by adding Dox and PI, for the Env assay by staining for HA, or, importantly, for both by adding Dox, PI, and HA staining.
The mixing experiment showing that each population kept the distinct characteristics of each assay gave us the confidence to proceed and engineer a two-assays-one-cell system. This multiplexed one-cell system platform represents a useful approach for determining compound specificity when screening for inhibitors of various viral targets, as shown here for HIV-1 PR and Env. Furthermore, each assay functions as an internal control for compound specificity for the other, allowing for more selective targeted screening, all within the same cellular content. This platform can serve as a useful tool for targeted drug discovery as it allows monitoring multiple proteins/targets from the same virus simultaneously. This is the first cellular platform of its type that stably expresses two distinct assays that interrogate two processes that occur in two different cellular compartments.
Materials and Methods
Transfections
HEK 293T cells were plated in a 12-well plate at 150,000 cells per well 24 hours prior to transfection. Cells were then co-transfected with 350 ng of reverse tetra-cycline transactivator (pBMN-rtTA-i-Lyt2), UAS (pH-5X UAS-eGFP), and HIV protease/Gal4 fusion (pH-TRE-Gal4 PR) using high-potency polyethylenimine (PEI) (linear, MW 25,000; Polysciences, Inc) at 8 µL/µg of DNA diluted according to manufacturer's specification. Cells were then treated with 1 µg/mL Dox and 1 µM of the HIV PR inhibitor DAR when needed, and analyzed 48 hours posttransfection via flow cytometry.
Cell Maintenance
Phoenix GP cells and human embryonic kidney HEK 293T cells were maintained at 37°C and 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM; CellGro) supplemented with 10% fetal calf serum (FCS), penicillin–streptomycin, and 2 mM L-glutamine. Human T-cell line SupT1 was maintained at 37°C and 5% CO2 in complete Roswell Park Memorial Institute medium (RPMI) 1640 (CellGro) supplemented with 10% FCS, penicillin-streptomycin, and 2 mM L-glutamine. Phoenix GP cells were provided by Gary Nolan from Stanford University. HEK 293T cells were provided by Chris Glembotski from San Diego State University. SupT1 cells were obtained from the American Type Culture Collection (ATCC).
Antibodies and Reagents
Anti-FLAG antibody was obtained from Sigma-Aldrich. Anti-HA, anti-mouse IgG Alexa Fluor647, and anti-rabbit Alexa Fluor 647 were obtained from Cell Signaling. DCK was obtained from Tocris Bioscience and was dissolved in 10% dimethyl sulfoxide. Dox was obtained from Sigma-Aldrich and DAR from the National Institutes of Health (Bestesda, MD).
Generation of Infectious Viral Particles
For production of Moloney murine leukemia virus (MLV)-based retrovirus, the Phoenix GP cell line at 60%–70% confluence in a 10 cm2 plate was transfected with 3 µg of the transfer vector (pBMN-HIV Env wt-i-Blasticidin) and 3 µg of a vector expressing the envelope glycoprotein of the vesicular stomatitis virus (pCI-VSVg). Plasmids were mixed in 500 µL of FCS-free DMEM with 48 µL of PEI (8 µL per µg of DNA). DMEM media was replaced 24 hours posttransfection, and viral supernatant was collected at 48 and 72 hours after transfection. All viral stocks were filtered with 0.45 µm polyvinylidene fluoride (PVDF) filters (Olympus Manufacturers) and either used upon collection or frozen at –-80°C in 2 mL aliquots.
Transductions
SupT1 cells grown in RPMI at 1,000,000 cells/well in a six-well plate were prepared for transduction. Cells were treated with 4 µg/mL Polybrene (hexadimethrine bromide, Sigma-Aldrich) and transduced with viral stocks by hanging bucket rotors centrifuge (Becton Dickinson) at 1500 G, for 80 minutes at 32°C. Cells were then analyzed for expression 72 hours postinfection.
Flow Cytometry and Sorting
The Flow Cytometry Facility at SDSU performed analysis of cells on BD FACSAria at 488 and 633 nm lasers, as well as the BD FACSCanto using only the 488 blue laser and the 633 red laser. Data were collected using FACSDiva 6.1.1 software and analyzed by FlowJo version 6.7.1.
Author Contributions
Conceived and designed the experiments: DA, CAS, and RW. Analyzed the data: DA, CAS, and RW. Wrote the first draft of the manuscript: DA. Contributed to the writing of the manuscript: DA and RW. Agreed with manuscript results and conclusions: DA, CAS, CWR, and RW. Jointly developed the structure and arguments for the paper: DA and RW. Made critical revisions and approved the final version: DA and RW. All the authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
We would like to thank Dr. Garry Nolan from Stanford University for providing the Phoenix GP packaging cell line for the production of retroviral particles. We would also like to thank the Flow Cytometry Core Facility at San Diego State University for their service.